scRNA-seq_v3用户手册
scRNA-seq_v3用户手册
1. 第一章 应用范围
scRNA-seq_v3可以处理以下试剂盒的高通量测序数据分析:
DNBelab C系列高通量单细胞RNA文库制备试剂盒套装V3.0。
2. 第二章 产品介绍
2.1 分析流程图
scRNA-seq标准分析流程是基于DCS智能云平台开发的一款自动分析流程,其中包括数据过滤及质控、参考基因组比对和注释、有效细胞筛选、下游分析,以及报告输出等多个功能,图2-1是分析整体流程图:
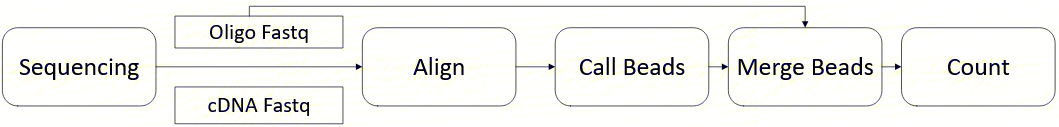
2.1.1 数据过滤及质控
根据配置文件中填入的数据质控标准进行fastq数据质控,经过过滤去除低平均碱基质量reads,截取指定长度reads等步骤,留下高质量的序列用于后续分析。
2.1.2 参考基因组比对和注释
使用STAR软件将过滤后的cDNA fastq文件与参考基因组进行比对,并进行UMI矫正,给出注释结果。
2.1.3 有效细胞筛选
采用EmptyDrops方法对原始矩阵进行有效细胞的选取,并通过余弦相似度合并细胞,生成用于下游分析的标准矩阵文件。
2.1.4 下游分析
基于生成的矩阵进行细胞质控和双胞过滤,对矩阵进行降维、聚类,进行细胞类型注释。
2.1.5 报告输出
综合分析结果,整理汇总成HTML报告。
3. 第三章 使用说明书
scRNA-seq标准分析流程通过STOmics时空云平台系统进行样本输入及报告输出的全流程管理。下面具体介绍基于STOmics时空云平台系统使用scRNA-seq标准分析流程的操作指南。
3.1 指南概述
本章介绍如何使用scRNA-seq标准分析流程进行分析。在使用之前,请认真阅读并理解其中内容,保证能够正确使用scRNA-seq。
3.2 使用场景一:手动投递
操作共包括四个步骤:上传数据、构建参考基因组(可选)、样本信息录入、启动分析。完成样本信息录入后,运行任务,当客户在看到任务状态为completed时,代表任务已完成,即可查看报告部分(详见3.4部分)。
3.2.1 步骤一:上传数据
- 点击导航栏**【数据管理】,进入数据管理页面,进入目标文件夹,点击右上角【+添加文件】-【工具上传】**上传数据(图3-1)。
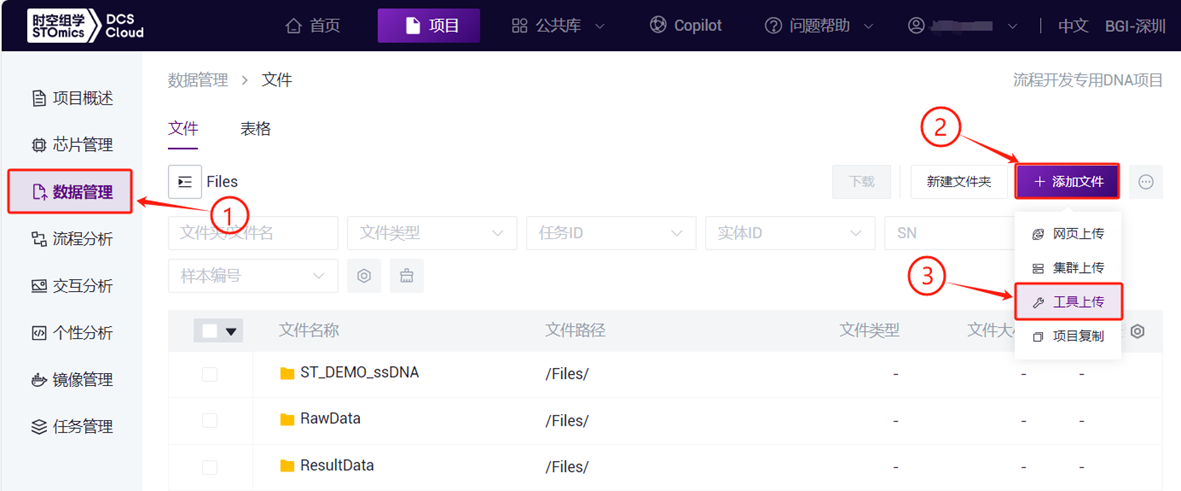
- 点击**【立即上传】浏览并选择所需文件(图3-2),上传完成后文件会在目标文件夹中显示(如首次上传,则需点击【安装并启动传输客户端】**安装所需工具)。

3.2.2 步骤二:构建参考基因组(可选)
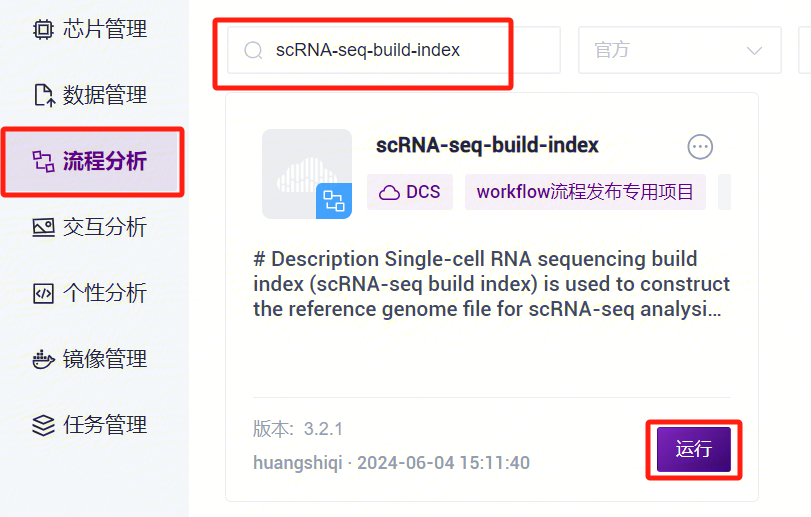
- 点击导航栏**【流程分析】进入流程分析页面,在搜索框输入scRNA-seq-build-index**,点击**【运行】**(图3-3)。
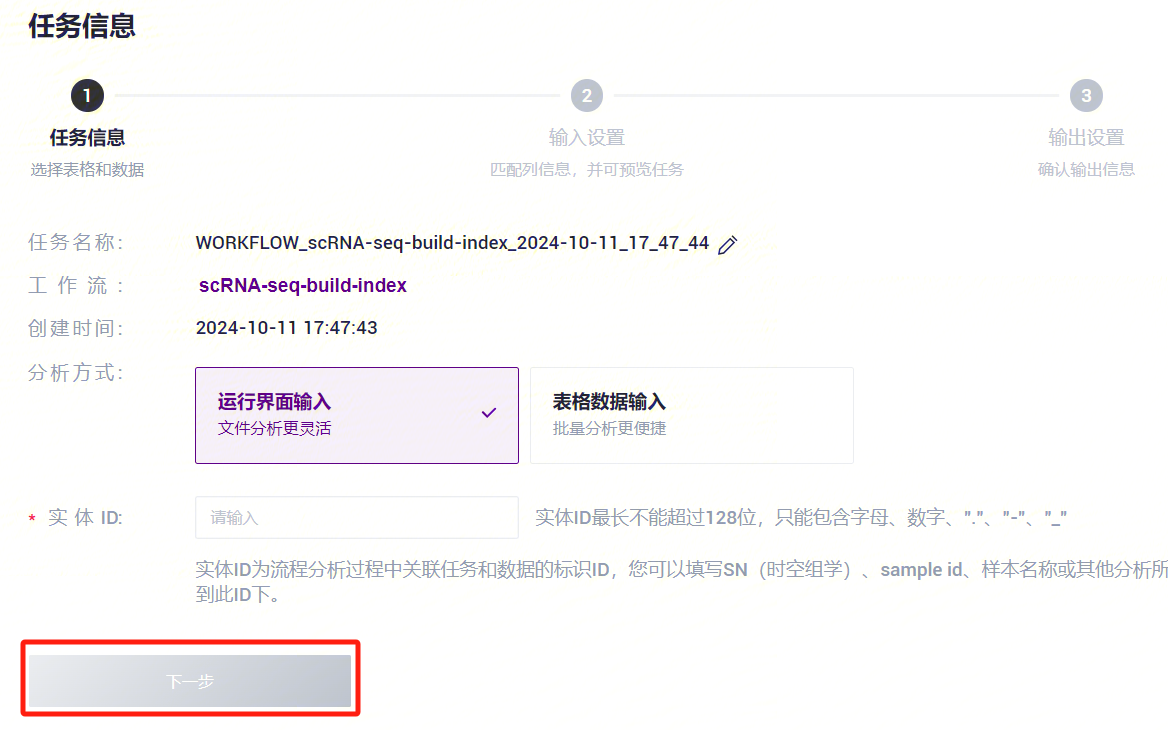
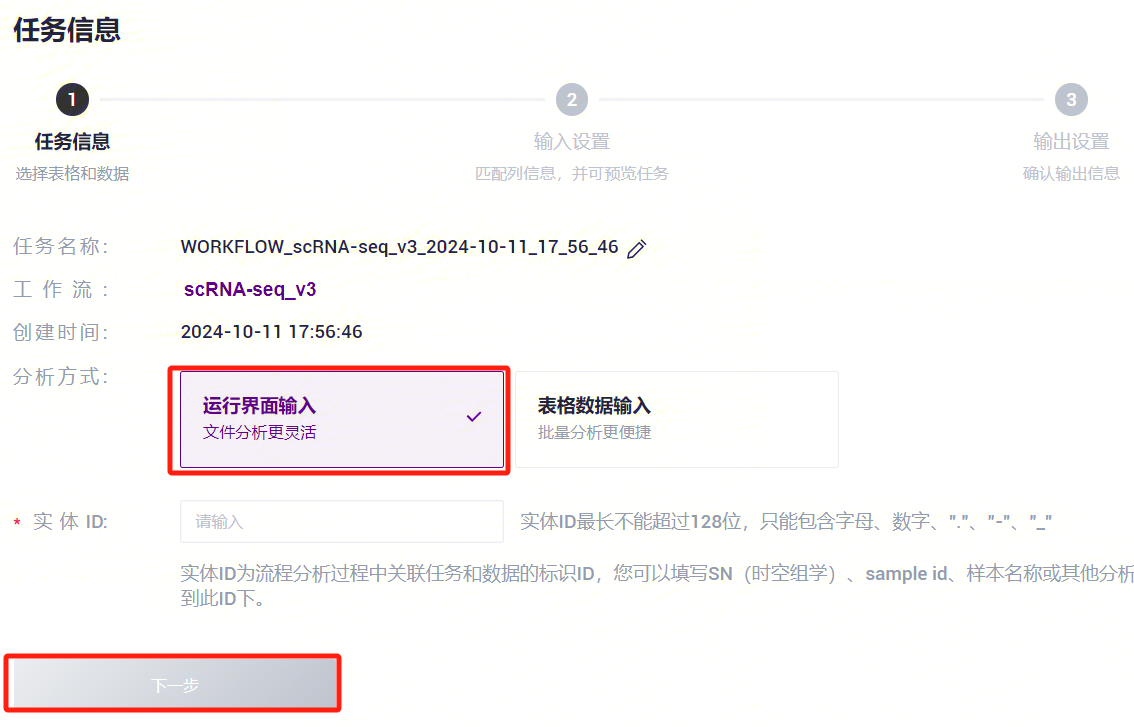
- 选择运行界面输入,输入Entity ID,点击**【下一步】**(图3-4)。
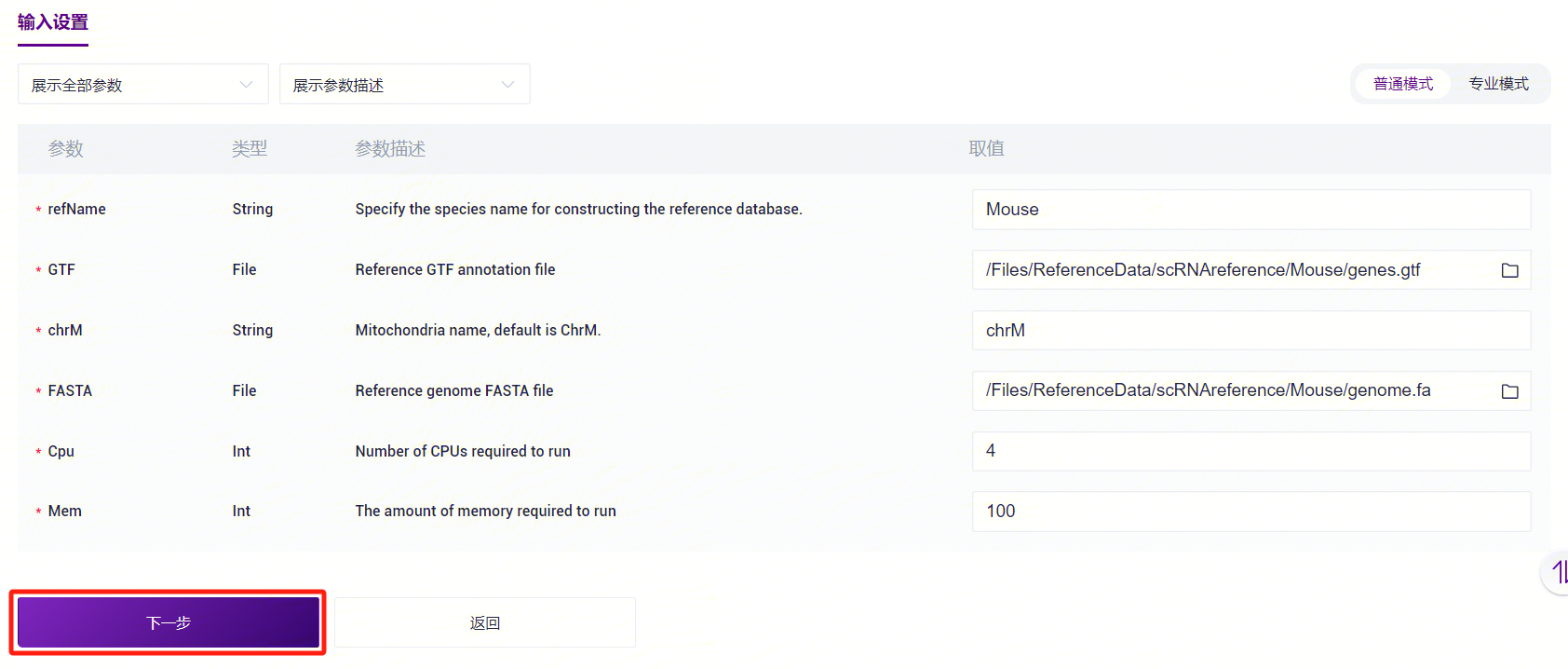
- 录入参考基因组信息,完成后点击**【下一步】**(图3-5)。
注意
参考基因组信息录入变量说明:
refName:参考基因组的物种名称,会显示在分析报告中;
GTF:基因组注释GTF文件;
chrM:线粒体名称;
FASTA:基因组FASTA文件;
Outdir:输出文件路径;
Cpu:运行所需CPU;
Mem:运行所需内存大小。
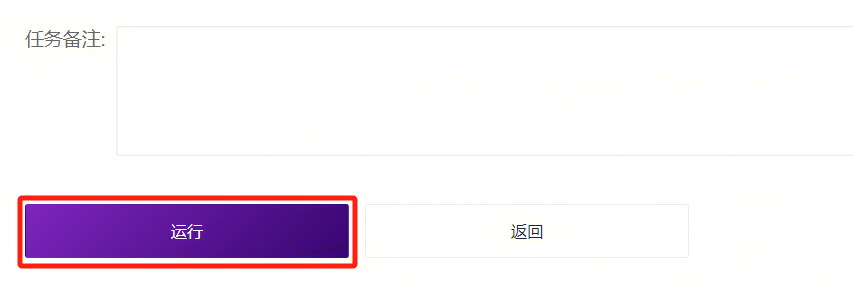
- 点击**【运行】**启动分析(图3-6)。
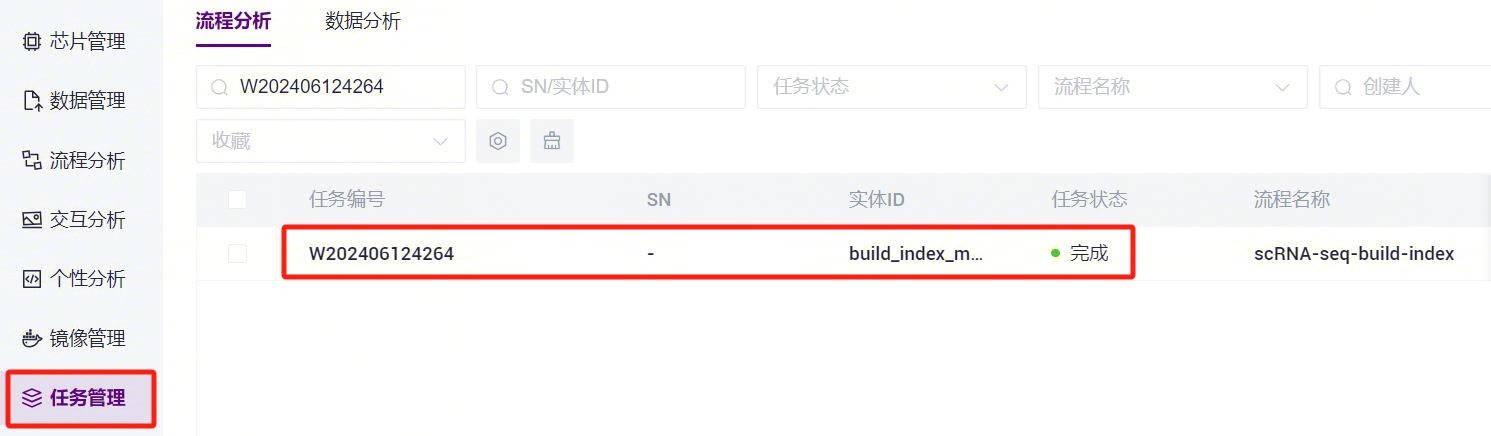
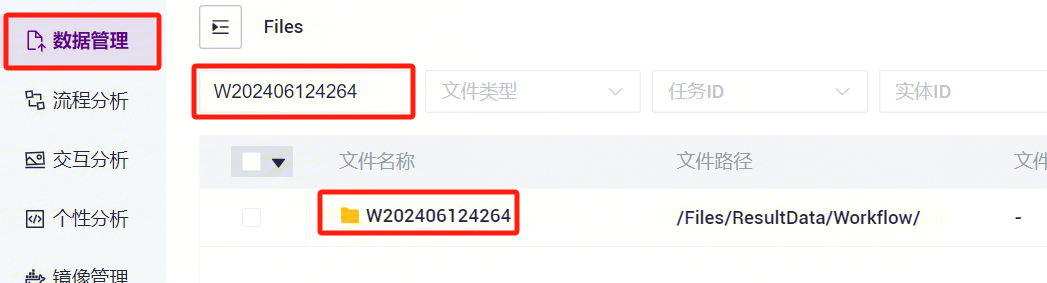
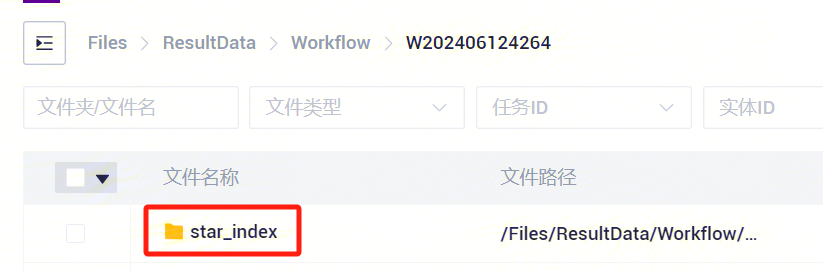
- 任务完成后,Status显示为completed,复制Task ID(图3-7),点击导航栏**【任务管理】,输入Task ID进行搜索(图3-8),其中star_index**文件将作为参考基因组文件参与单细胞RNA标准流程分析(图3-9)。
3.2.3 步骤三:样本信息录入
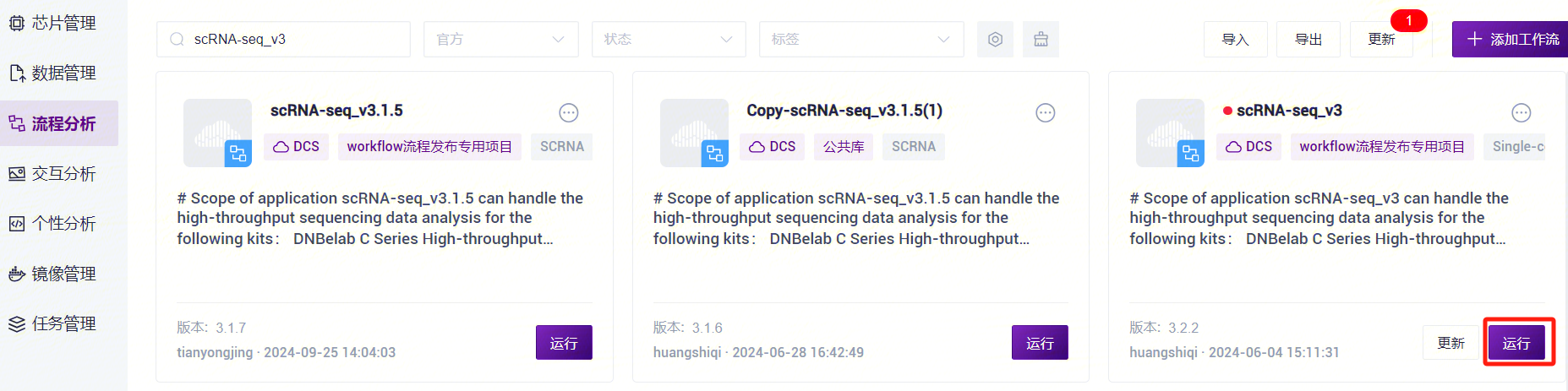
- 点击导航栏**【流程分析】进入流程分析页面,在搜索框输入scRNA-seq_v3****,点击【运行】**(图3-10)。
- 选择运行界面输入,输入Entity ID,点击**【下一步】**(图3-11)。
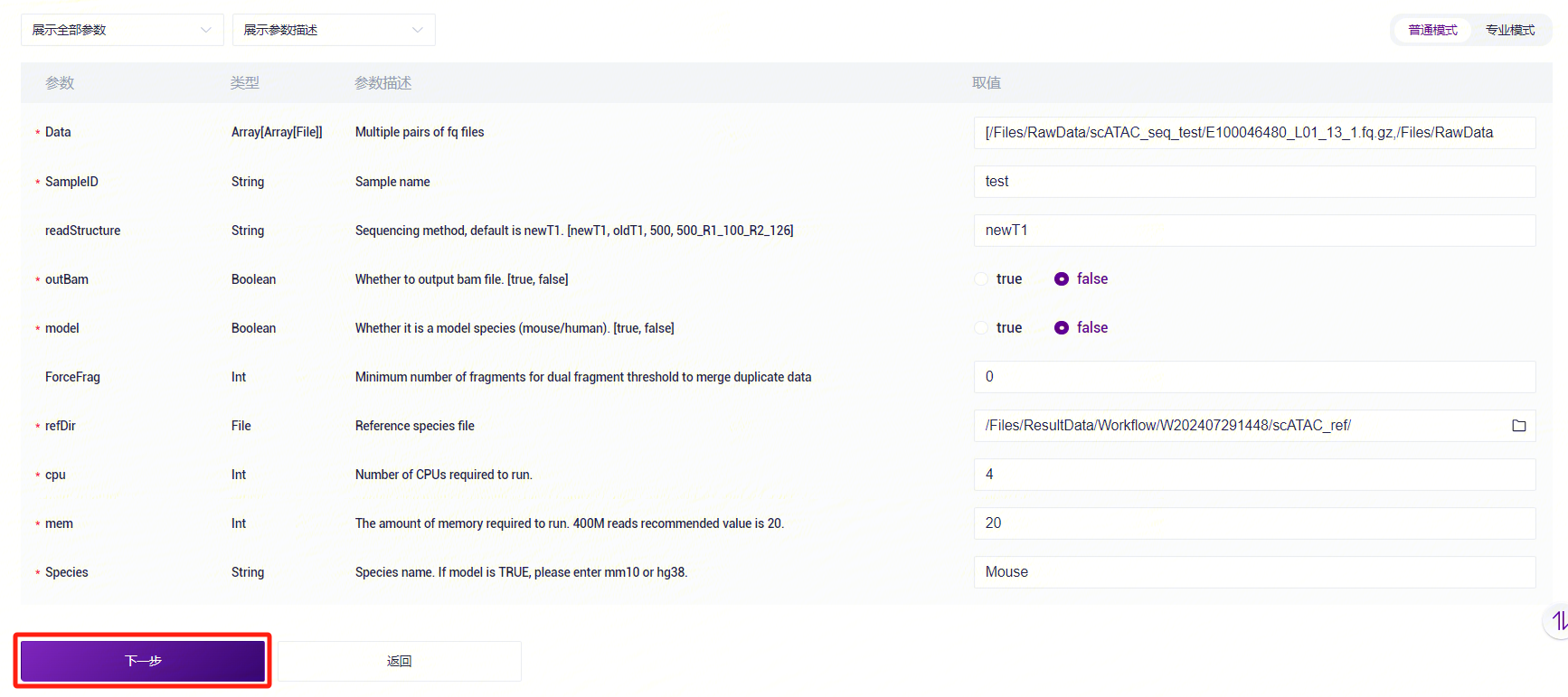
- 录入样本信息,完成后点击**【下一步】**(图3-12)。
注意
样本信息录入变量说明:
sampleID:样本名称,默认与Entity ID一致;
cDNA:cDNA文库fastq格式的R1、R2端序列;
Oligo:oligo文库fastq格式的R1、R2端序列;
genomeDir:参考基因组文件,如在步骤二(3.2.2 构建参考基因组)自主构建,选择Task ID文件夹内的star_index;
Outdir:输出文件路径;
expectcells:期望的细胞数量,可根据经验自行选择细胞数目;
forcecells:强制选取beads数目,根据骤降图强制截取 beads 数目;如为0则不参与计算;
Cpu:运行所需CPU大小,所需CPU最低值为8,推荐值为16;
Mem:运行所需内存大小。
3.2.4 步骤四:启动分析
点击**【运行】**启动分析(图3-13)。
3.3 使用场景二:表格投递
操作共包括五个步骤:上传数据、构建参考基因组(可选)、下载样本模板、填写并导入样本模板、启动分析。完成样本模板导入后,可批量运行任务,当客户在看到任务状态为completed时,代表任务已完成,即可查看报告部分(详见3.4部分)。
3.3.1 步骤一:上传数据
与使用场景一(手动投递)步骤一一致(详见3.2.1步骤一:上传数据)。
3.3.2 步骤二:构建参考基因组(可选)
与使用场景一(手动投递)步骤二一致(详见3.2.2步骤二:构建参考基因组)。
3.2.3 步骤三:样本信息录入表格下载
- 点击导航栏**【数据管理】,选择【表格】-【下载模板】(如图3-14), 点击【Data model template】**,选择scRNA-seq_v3.2.0模板下载。
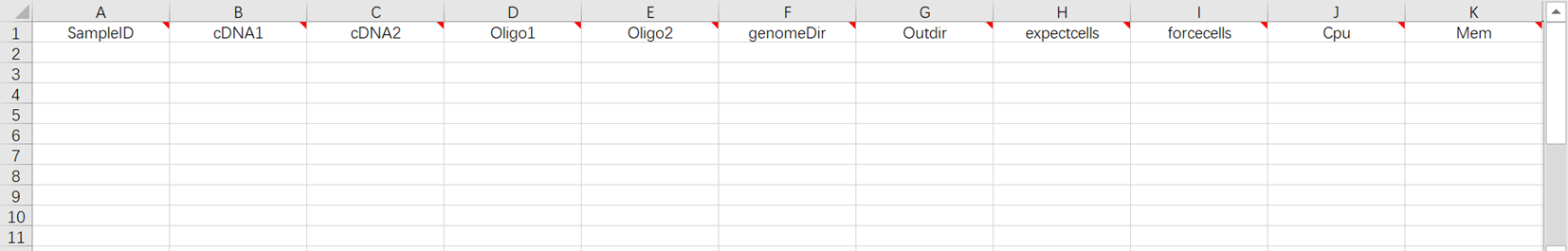
- 打开后scRNA-seq_v3.2.0样本模板Excel如图3-15。
3.3.4 步骤四:样本信息导入
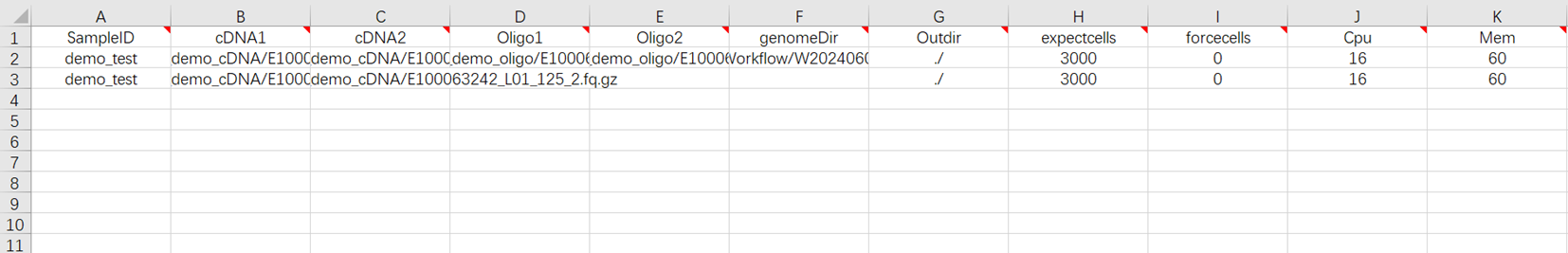
- 该使用场景条件下,样本导入表格需填写工作表(图3-15)。该场景表示对已测序完成的样本数据在导入表格后,直接进入分析。
注意
注意事项:
[1] 导入文件路径必须在云平台已存在。
[2] 模板中,所有内容均为必填项,任何字段不得为空。
[3] Excel中的SampleID需唯一,如SampleID一致则默认为同一个任务,合并多对cDNA的fq进行分析,适配cDNA补充测序的情况。
[4] Excel中不能合并单元格,单元格内容前后不能有空格或特殊字符。
[5] 分析样本录入(图3-16):
sampleID:样本名称,默认与Entity ID一致;
cDNA:cDNA文库fastq格式的R1、R2端序列;
Oligo:oligo文库fastq格式的R1、R2端序列;
genomeDir:参考基因组文件,如在步骤二(3.3.2 构建参考基因组)自主构建,选择Task ID文件夹内的star_index文件;
Outdir:输出文件路径;
expectcells:期望的细胞数量,可根据经验自行选择细胞数目;
forcecells:强制选取beads数目,根据骤降图强制截取 beads 数目;如果是0保留全部细胞;
Cpu:运行所需CPU大小,所需CPU最低值为8,推荐值为16;
Mem:运行所需内存大小。
- 配置好样本模板的分析样本录入工作表后,回到**【数据管理】界面,点击【表格】-【+新增表格】**(图3-17)。
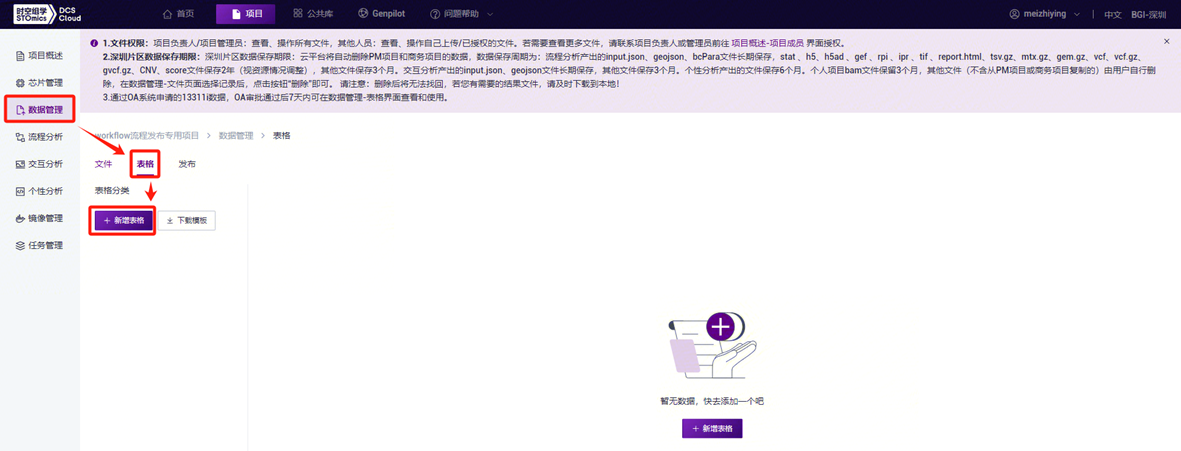
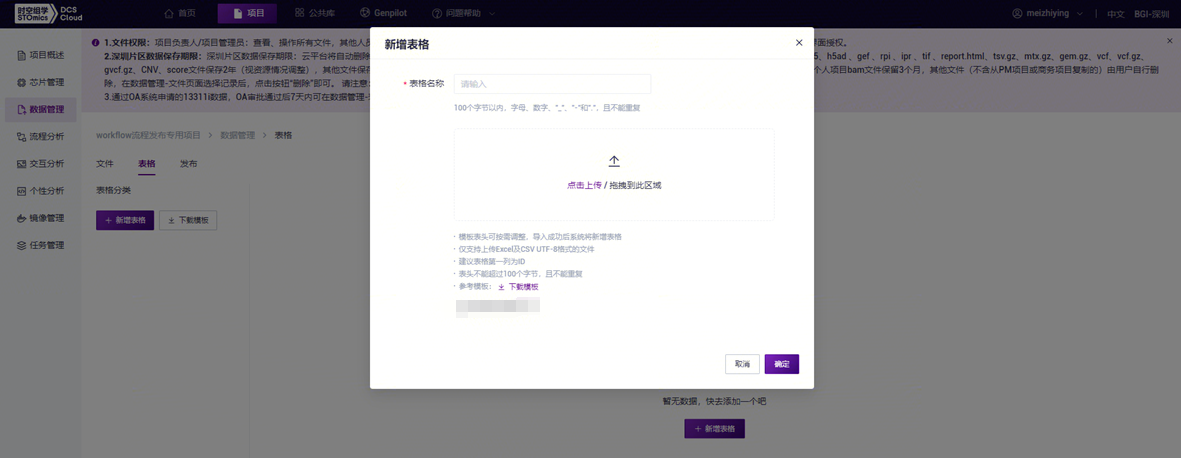
- 点击**【点击上传/拖拽到此区域】浏览并选择已填好样本信息的表格,点击【确定】**(图3-18),上传完成后文件会在目标文件夹中显示。
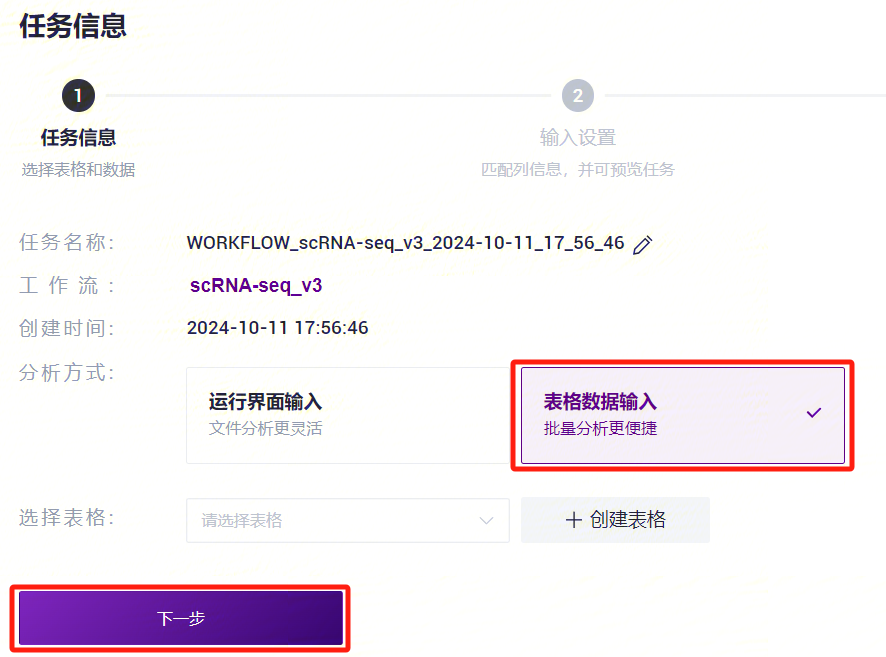
- 点击导航栏**【流程分析】进入流程分析页面,在搜索框输入scRNA-seq_v3****,点击【运行】**(图3-19)。

- 选择表格数据输入,点击Please select table处,选择在本小节3)中导入的表格,选中所需的行,点击**【下一步】**(图3-20)。
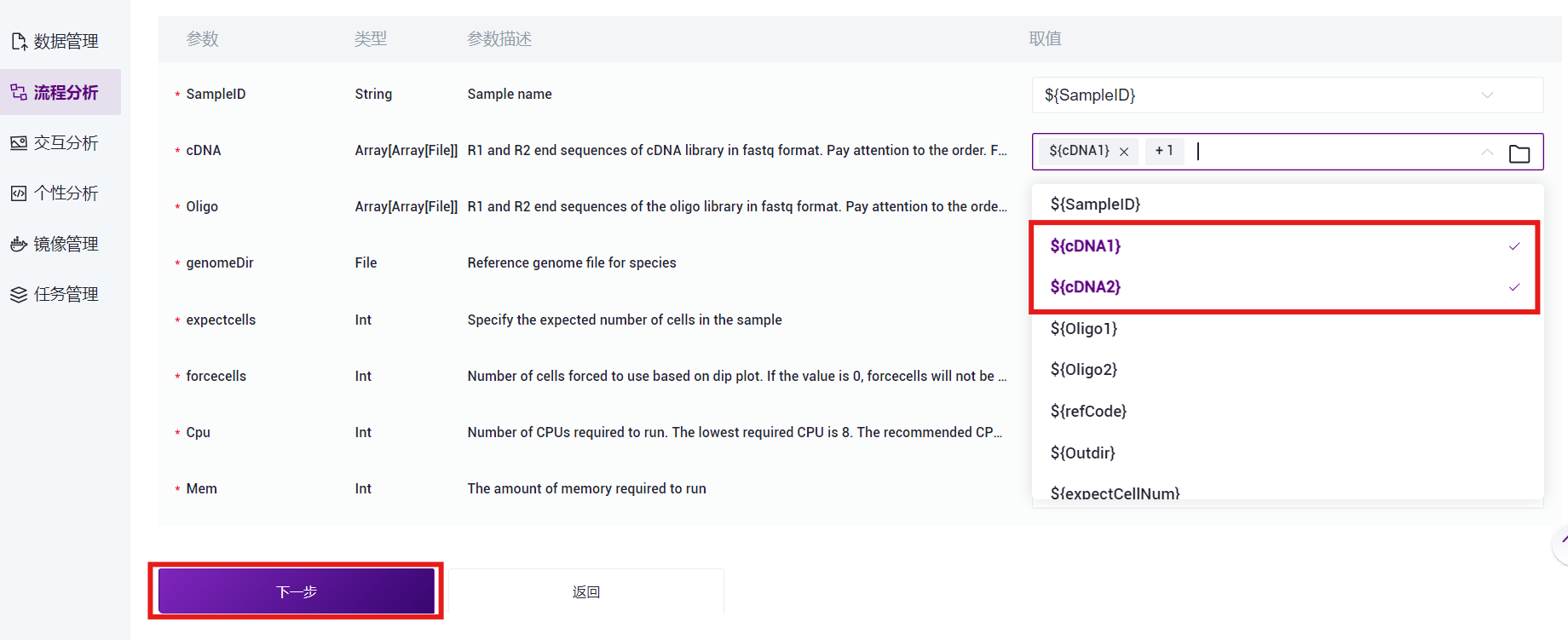
- 在Values处点击并选择对应的值,如cDNA选取${cDNA1}和${cDNA2},注意按顺序选取(如图3-21);Oligo选取${Oligo1}和${Oligo2},注意按顺序选取(如图3-22)。

- 录入样本信息,完成后点击**【下一步】**,确保参数设置无误(如图3-23)。
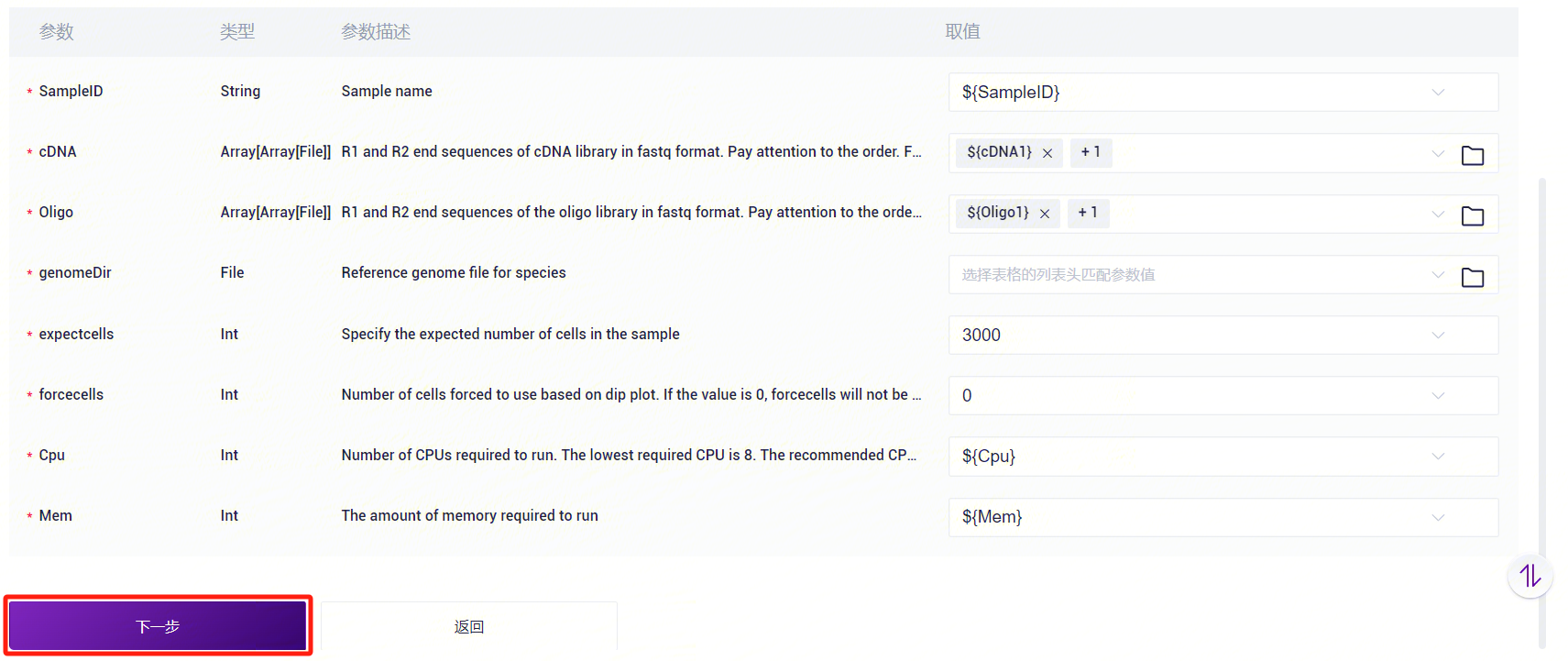
3.3.5 步骤五:启动分析
点击**【运行】**启动分析(图3-24)。
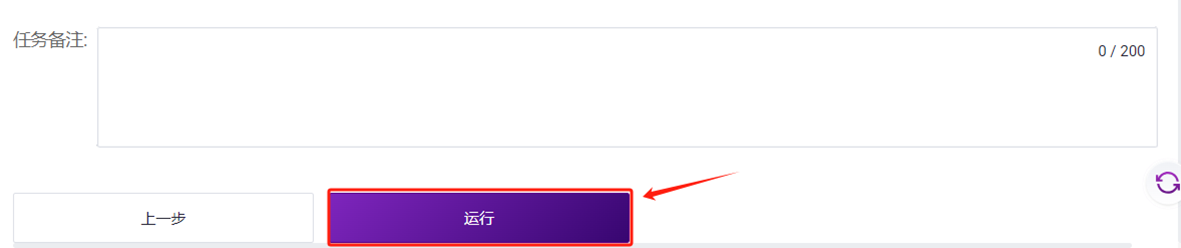
3.4 报告查看及结果文件下载
- 点击导航栏**【任务管理】**,当任务状态显示为completed时,代表任务已完成,即可查看报告部分(如图3-25)。
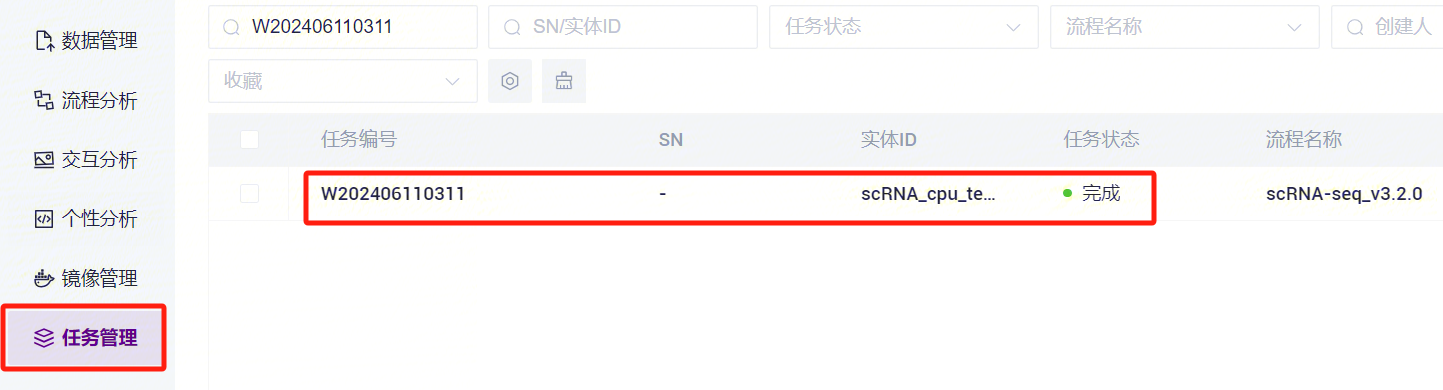
- 点击导航栏**【数据管理】,进入数据管理页面,根据任务的Task ID进行搜索,点击进入Task ID**文件夹(如图3-26)。
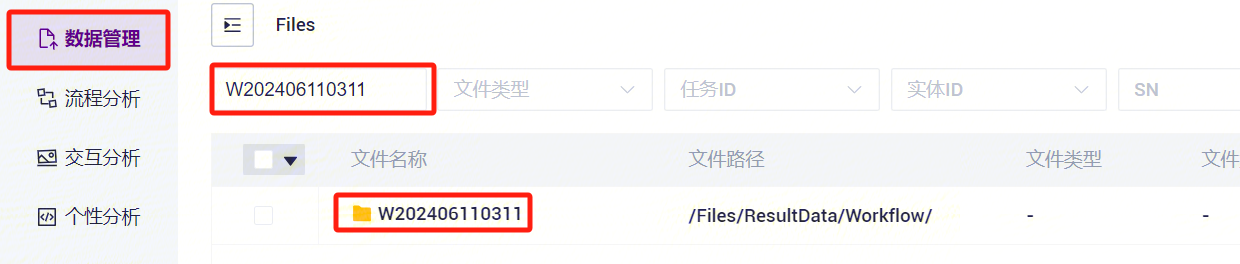
- 点击进入Entity ID文件夹(如图3-27)。
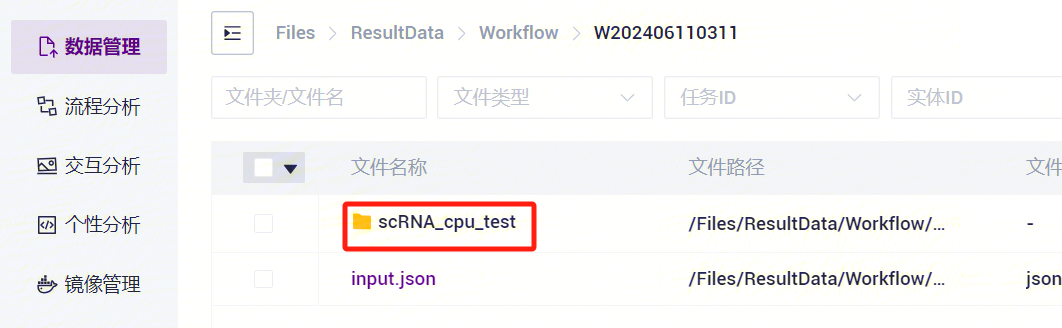
- 点击进入04.report文件夹(如图3-28)。
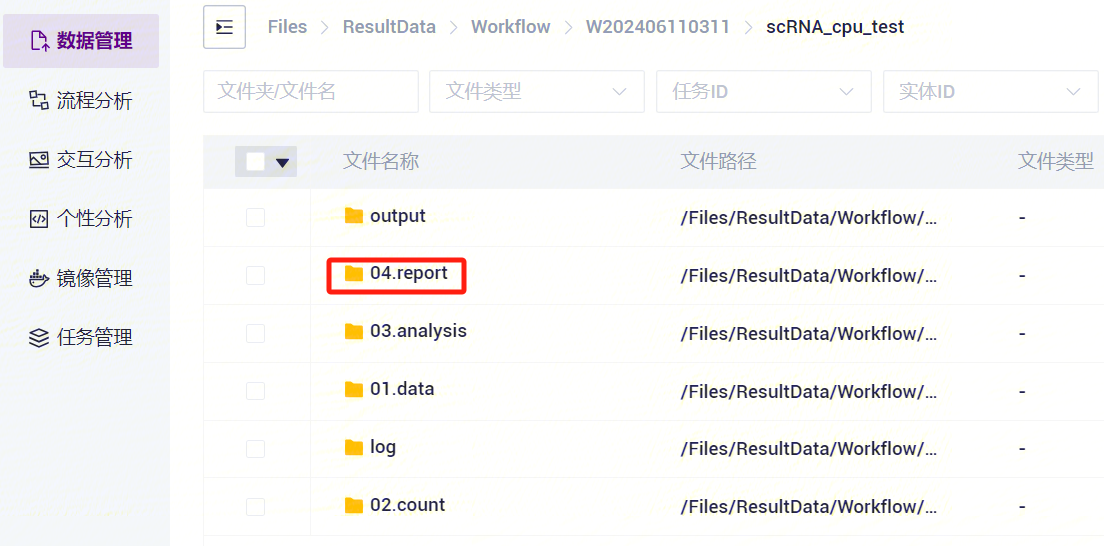
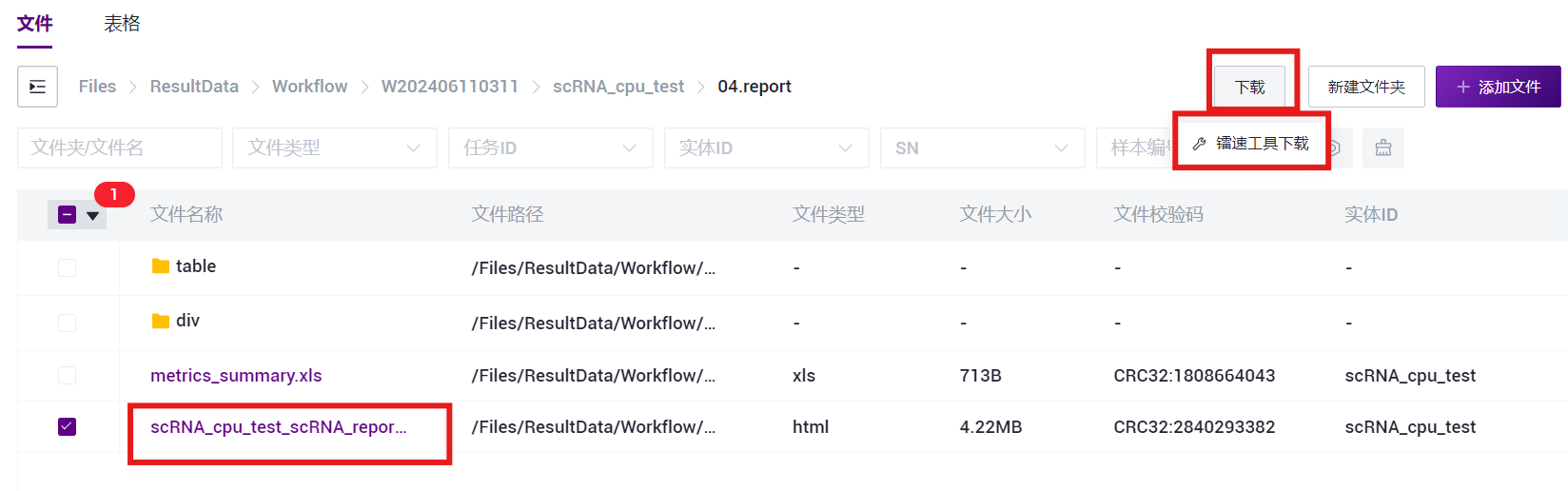
- 选中Entity ID_report.html文件夹,点击**【传输】-【下载】-【镭速工具下载】**(如图3-29)。
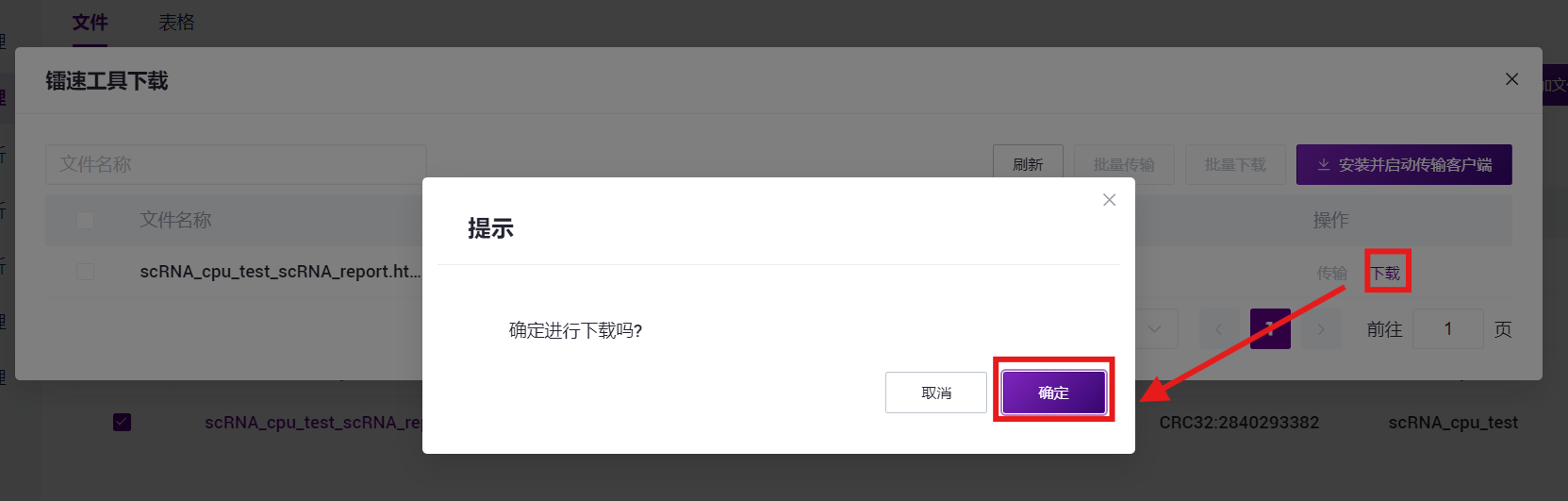
- 点击**【传输】-【下载】-【确定】**,选择目标目录并下载报告(如图3-30)。
4. FAQ
- 官方的单细胞流程有哪些,分别对应试剂盒的版本是什么?
scRNA-seq_v3与scRNA-seq-3.1.5为官方维护流程。


scRNA-seq-3.1.5,对应试剂盒DNBelab C系列高通量单细胞RNA文库制备试剂盒套装V2.0。
scRNA-seq_v3对应智造的流程是dnbc4tools v2.1.2,对应试剂盒:DNBelab C系列高通量单细胞RNA文库制备试剂盒套装V3.0。
- 构建Reference后gtf文件格式要求是什么?
gtf文件中染色体名称与基因组文件中的染色体名称要一致。