此功能的使用需根据SAW版本进行区分。
SAW >= v8.0:
①使用SAW makeRef构建比对所需的参考基因组索引文件,若需进行rRNA去除,需要在构建时设置--rRNA-fasta参数,自动添加rRNA信息并构建索引文件。
②运行SAW count时开启--rRNA-remove参数即可,并且使用添加了rRNA信息的索引文件。
③去除rRNA的步骤细节和原理详见《SAW 用户手册》>使用教程>比对索引构建>#SAW makeRef>##转录组比对>###添加 rRNA 信息至参考基因组。
SAW < v8.0:
①可以在参考基因组FASTA文件中手动添加rRNA序列,重新构建reference index后,再在SAW运行mapping时打开rRNAremove开关,过滤掉比对上rRNA序列的reads。rRNA过滤功能在SAW v6.0版本新增。
②添加rRNA序列规则:在FASTA文件中增加待过滤的rRNA序列,“>”开头的序列名称需要在第一个部分的名称最后增加“_rRNA”,用于程序识别。示例如下:

in=<mask>
in1=<lane_read_1.fq.gz>
in2=<lane_read_2.fq.gz>
barcodeReadsCount=<lane.barcodeReadsCount.txt>
barcodeStart=0
barcodeLen=25
umiStart=25
umiLen=10
umiRead=1
mismatch=1
bcNum=<CIDCount>
polyAnum=15
mismatchInPolyA=2
rRNAremov
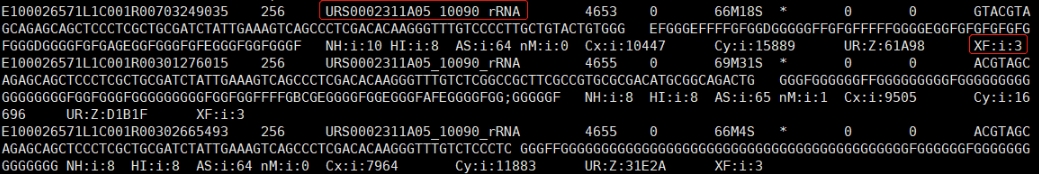
④如果query read比对上rRNA,其比对记录第三列RNAME为参考基因组中添加了"_rRNA"尾缀的序列名称,第十二列Optional fields的XF:i标签为3。后续注释时,根据XF标签的记录,统计rRNA的比例。