空间转录组学通过整合分子活动与精准定位空间信息,改变了研究者对基因表达调控机制的理解。作为这一领域的关键技术,华大时空组学技术Stereo-seq凭借卓越的单细胞分辨率、高通量芯片承载能力以及跨物种适用性,已成为解析复杂组织中基因表达模式的重要工具,为多组织类型和多尺度生物学研究提供了高效的空间转录组分析解决方案。
在Stereo-seq的关键应用场景中,空间基因共表达分析正成为解析局部微环境中功能基因网络的核心方法。这一方法有效连接了传统共表达分析与高分辨率空间定位需求,填补了传统基因表达研究与复杂组织空间异质性解析之间的技术鸿沟。
本文将深入探讨空间基因共表达的理论基础,系统评估其在Stereo-seq数据中的算法性能,并通过实际案例展示Stereo-seq在转化医学中的应用潜力。
— 评估结果抢先看 —
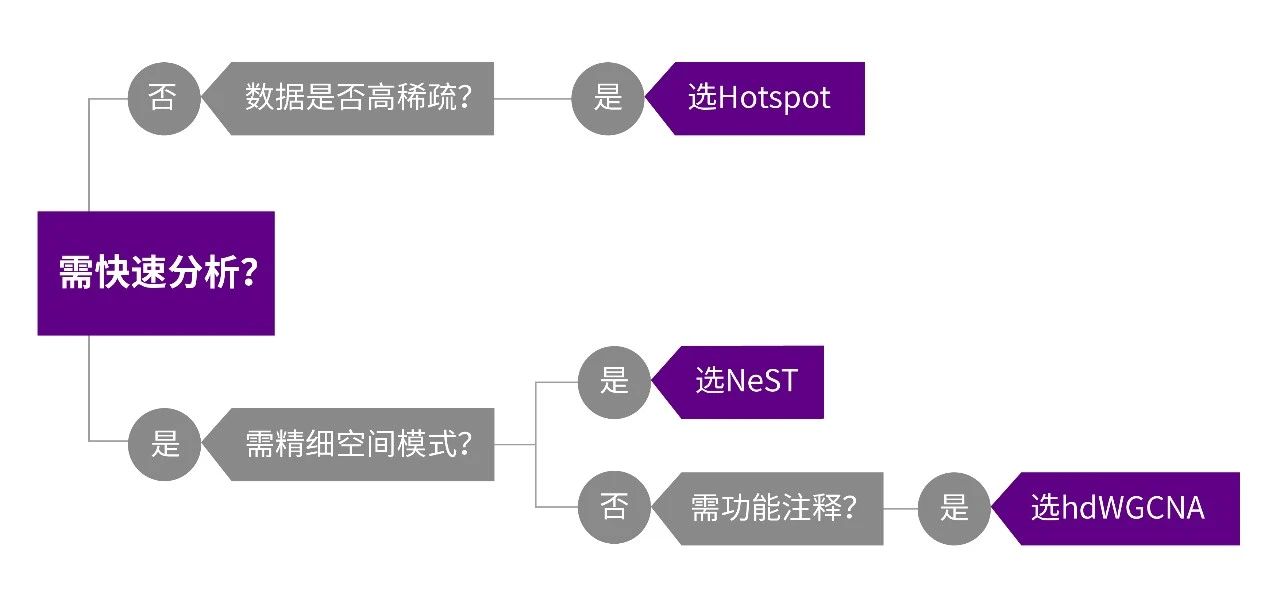
在选择空间共表达算法时,建议根据具体样本特征并且结合个性化参数调整。三种算法的性能表现与其理论基础高度契合:若需快速获得分析结果,可优先考虑hdWGCNA和NeST。其中,hdWGCNA在基因功能注释的生物学一致性方面表现更优,而NeST算法在空间模式的精准识别上具有明显优势。当面对高稀疏性的空间转录组数据矩阵时,Hotspot算法因其处理稀疏数据的能力而成为更优选择。

推荐等级以星级表示,三星表示最高推荐级别
什么是空间基因共表达?
空间基因共表达分析在传统方法(如加权基因共表达网络分析,WGCNA)基础上进一步拓展,通过整合空间位置信息构建基因共表达网络。与传统方法仅关注基因在跨样本表达模式中的协同变化不同,空间共表达特别聚焦于识别那些既呈现表达模式共变、又严格定位于特定组织微环境的基因模块(如图1所示)。这种空间维度的引入对解析基因功能受微环境调控的复杂系统(如肿瘤异质性、神经回路构建及器官发育)具有重要意义,能够揭示传统方法无法捕捉的区域性基因协同作用机制。
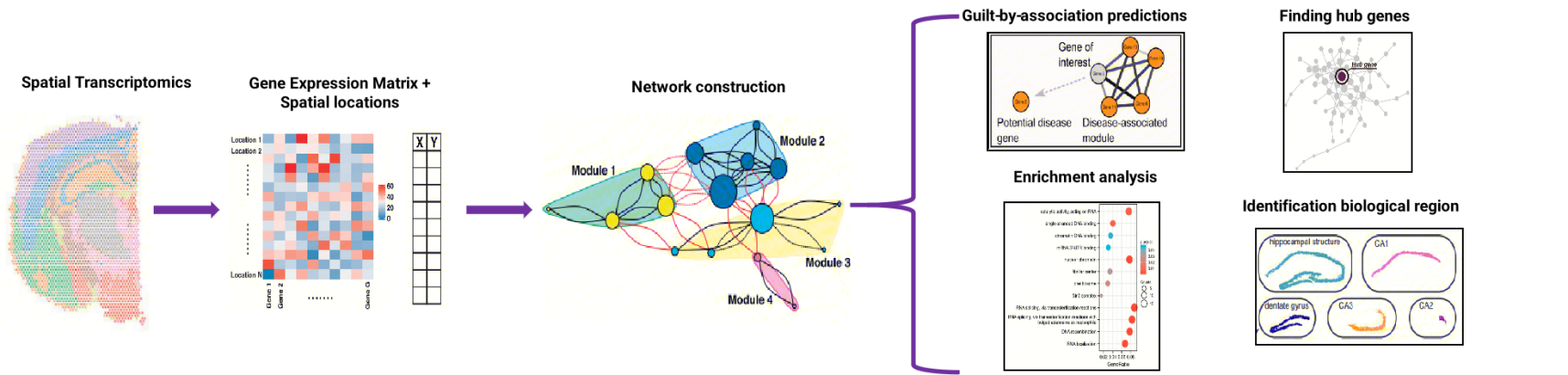
图1. 空间基因共表达分析流程
该算法通过整合基因表达水平与空间位置信息,量化基因间的关联性,进而解析基因相互作用网络,并识别具有协同表达特征的基因模块,以揭示空间维度下的基因协作机制。
我们选择了三种空间基因共表达算法——NeST、Hotspot和hdWGCNA——这些算法能够稳健整合空间信息,并与高分辨率数据集兼容,且具有以下优势:
▪ 无需依赖细胞类型注释,从而避免了因注释偏差导致的分析倾向性;
▪ 通过直接处理原始数据而非插补缺失表达值,有效保留了数据的完整性,确保了分析结果的可靠性。
01、NeST原理
▪ 密度聚类:采用DBSCAN的算法,通过对邻近空间点的聚类分组识别空间可变基因。
▪ 基于图的聚类:将具有协同表达模式的空间可变基因归集为功能相关的共表达模块。
▪ 优势:通过整合空间距离信息与基因表达数据,能够在组织样本的亚细胞层级分辨率上精确区分不同精细解剖结构,有效捕捉空间异质性特征。

图2. NeST算法的示意图1
02、Hotspot原理
▪ 基于KNN的图构建:利用空间坐标或谱系信息构建细胞相似性图谱。
▪ 统计检验:通过比较每个基因的表达与其相似性图谱(局部邻域)来识别空间可变基因。
▪ 层次聚类:根据基因两两相似性将具有协同空间表达模式的基因归类为功能相关的共表达模块。
▪ 优势:采用严格的统计框架,通过多重检验校正后的P值可有效控制假阳性率。

03、hdWGCNA原理
▪ 元细胞聚合:利用KNN将具有相似转录组特征的细胞聚类为"元细胞"。
▪ 加权邻接矩阵:计算元细胞间的基因共表达权重。
▪ 层次聚类:应用WGCNA识别共表达模块并识别具有协同表达特征的模块并解析其生物学功能。
▪ 优势:通过网络拓扑结构的稳健性分析,能够有效整合空间转录组与单细胞分辨率数据。
01、测试的Stereo-seq数据
我们共测试了11个样本,包括小鼠大脑和多种人类肿瘤样本。每个样本测试了cellbin、bin20、bin50和bin100(若可用)。测试数据集覆盖了bin20中基因数从约100到900的不同情况(见表1)。

表1. 测试Stereo-seq数据中不同bin大小的基因计数
02、评估方案
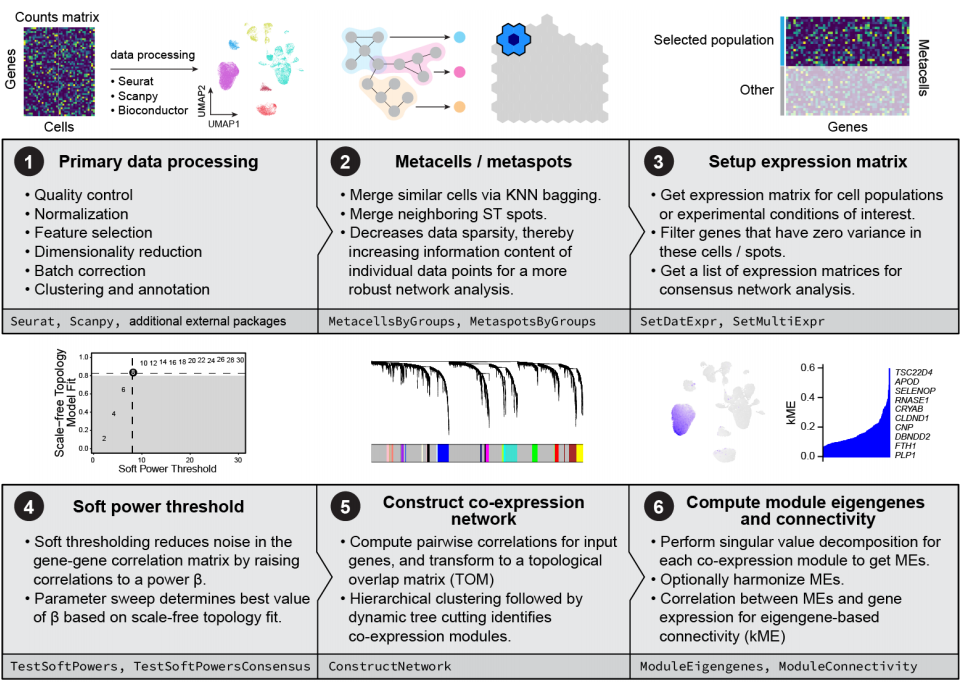
我们在相同计算条件下评估NeST、Hotspot和hdWGCNA,均限制为8个CPU核心并行处理。保留官方教程的默认参数,仅调整空间距离相关设置以匹配数据集的bin大小分辨率。性能评估包含四个关键维度共八项定量指标(表2)。
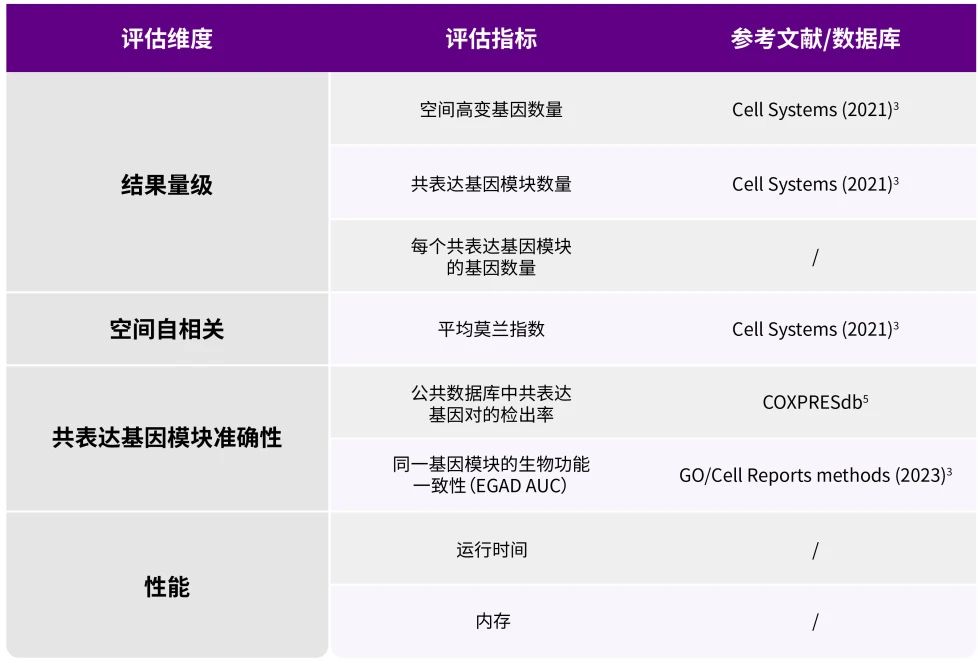
表2. 评估方案,主要关注涉及8个指标的四个方面,参考相关算法文章和公共数据库
详细评估结果
▪ 结果量级
关于空间基因共表达结果的量级(图5),Hotspot算法在识别空间高变基因和共表达模块的数量上展现出更优性能,显著优于NeST和hdWGCNA两种方法。具体而言,hdWGCNA呈现出特定的特征:其构建的基因模块数量相对较少(8-12个模块),但每个模块包含的基因数量显著更多(单个模块规模介于300至600个基因之间)。这一特点与Hotspot算法生成更多数量的模块形成鲜明对比,体现了不同算法在空间转录组数据解析策略上的差异。
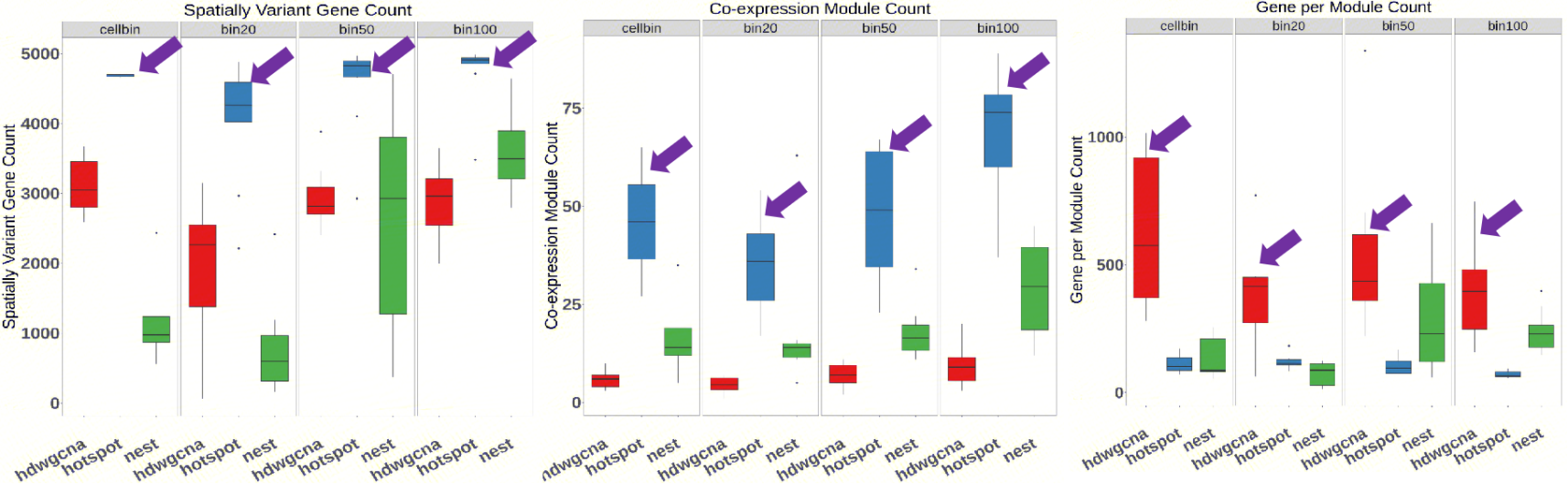
图5. 不同算法的结果规模。空间高变基因数量(左),共表达基因模块数量(中),每个共表达基因模块的基因数量(右)
▪ 空间自相关
关于空间自相关检测(图6),我们的分析表明NeST算法在cellbin和bin20数据集中显著优于Hotspot和hdWGCNA,能更好地富集空间一致性基因模式。然而,当空间分辨率逐渐降低时,NeST的性能优势呈现递减趋势。这种现象可能源于空间分辨率下降导致基因表达信息的减少,使得不同算法间的性能差异趋于模糊。

图6. 不同算法的空间自相关。计算平均莫兰指数以评估空间自相关。我们随机选取相同数量的空间高变基因和共表达基因集作为基线(浅色框),以便更直观地比较结果
▪ 共表达基因模块准确性
在共表达基因模块准确性方面(图7),我们的评估显示hdWGCNA表现优于NeST和Hotspot,体现在基因模块的功能一致性更高,且对已知共表达基因对的检测灵敏度更强。
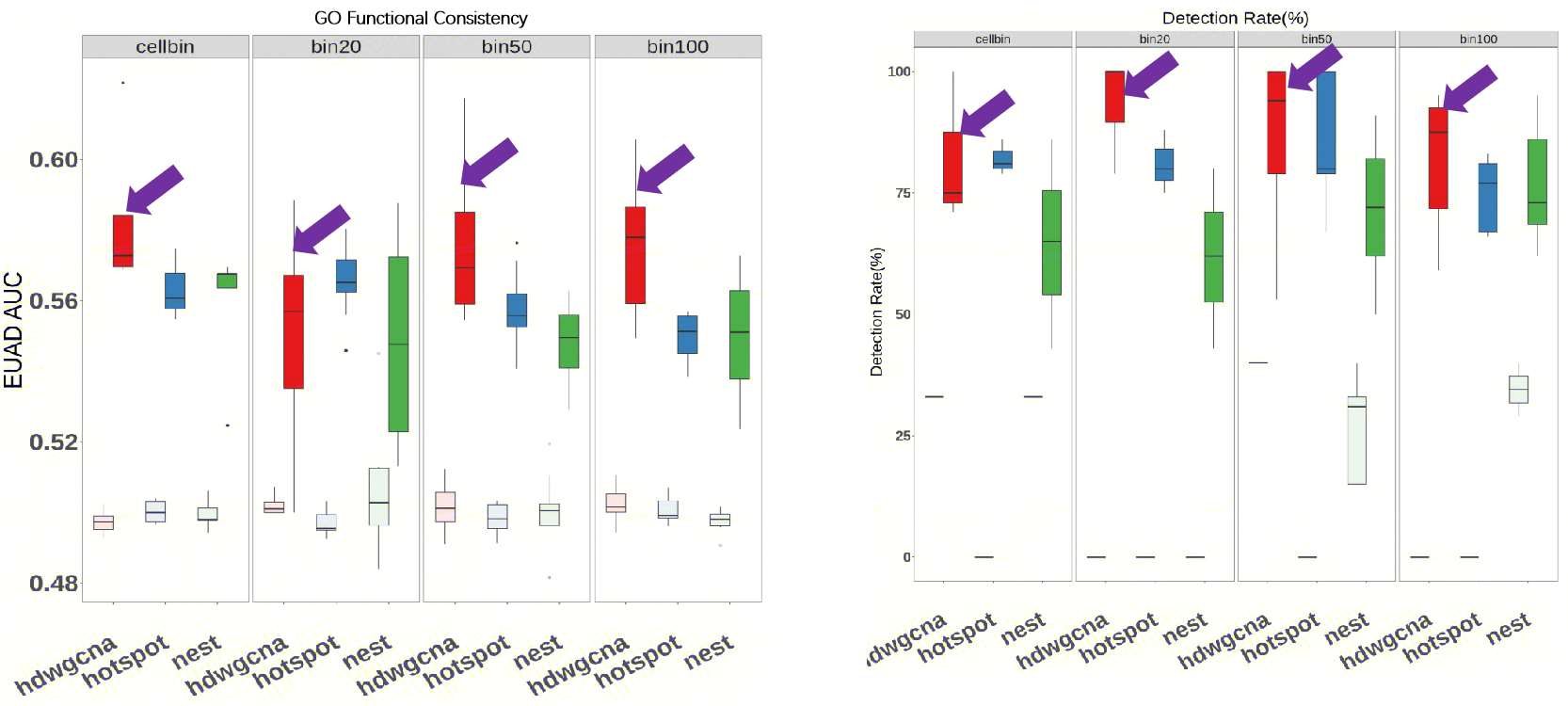
图7. 不同算法的共表达基因模块准确性。同一基因模块的生物功能一致性(左图EGAD AUC)。公共数据库中检测到的共表达基因对检出率(右图)
▪ 运行时间与内存占用
关于运行时间和内存分析,运行时间和内存需求与细胞及基因数量呈显著正相关,即数据规模越大,计算耗时和内存占用越高。Hotspot算法表现出最长的运行时间,而cellbin和bin20因数据集规模庞大处理时间显著延长(超过24小时),但Hotspot算法内存需求最低。hdWGCNA和NeST算法则展现出更优的计算效率:在bin50/bin100等较小规模数据集上仅需1-2小时即可完成分析,而处理cellbin/bin20这类更大数据集时,其运行时间也仅延长至3-5小时。
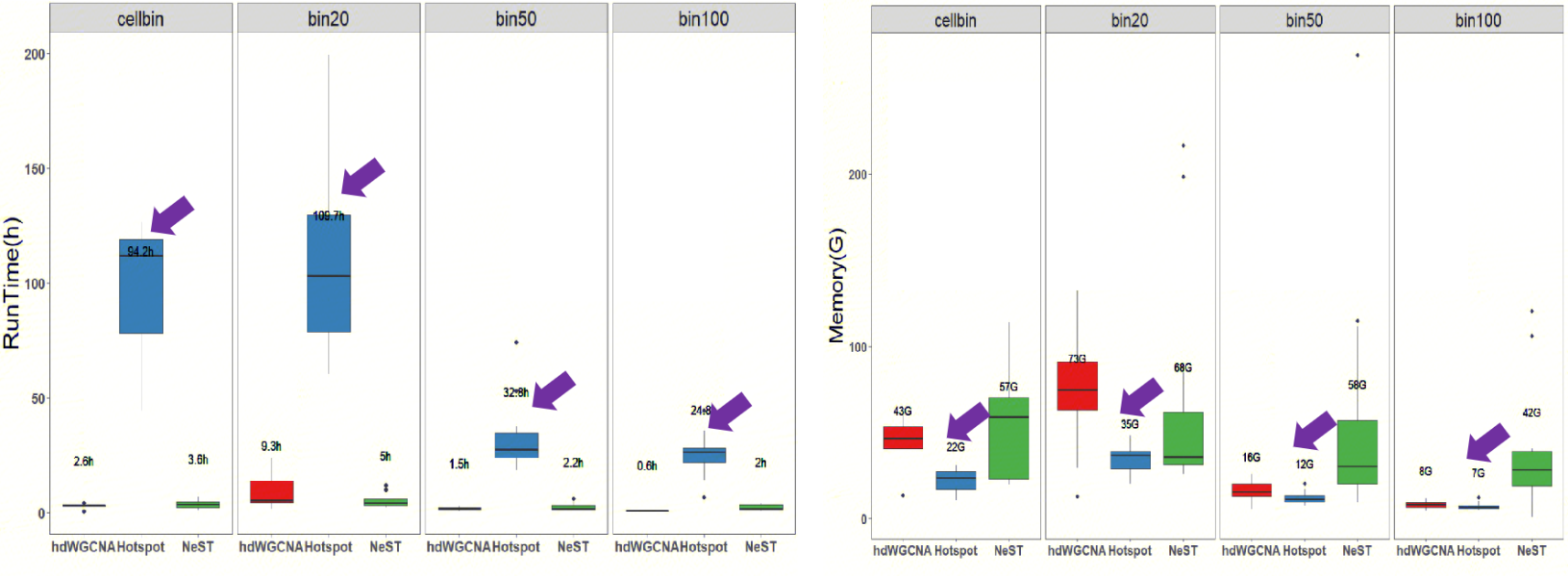 8. 不同算法的运行时间与内存使用情况
8. 不同算法的运行时间与内存使用情况
案例研究:将空间共表达转化为生物学见解
01、肿瘤亚型鉴定
我们旨在复现《Nature Genetics》论文中通过单细胞RNA测序(scRNA-Seq)鉴定肿瘤细胞亚群(MP6/MP7)并借助Stereo-seq技术展示其空间分布的研究成果。
本研究的核心目标是仅基于空间转录组学数据与基因共表达分析算法,无需依赖单细胞测序数据,独立实现肿瘤亚型的空间特征解析。通过三种独立算法的验证,本研究证实了证明仅用空间转录组学进行肿瘤亚型空间映射的可行性,为传统方法提供了更具成本效益的替代方案,同时通过基因模块的共表达模式挖掘,可进一步揭示肿瘤微环境的复杂调控网络(如原文图9所示)。
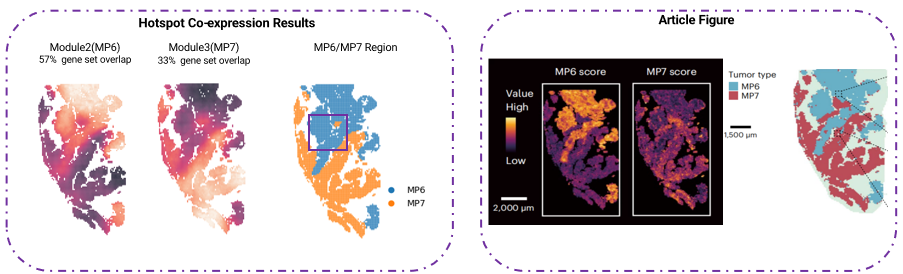
图9. Hotspot算法获得的结果与文献4中展示数据的对比
02、长链非编码RNA与信使RNA的共表达
利用时空转录组FFPE产品方案和NeST算法,我们系统分析了肺肿瘤样本中长链非编码RNA(MALAT1)与信使RNA(PTPN13)的空间共表达特征(图10)。通过结合细胞类型注释和空间聚类分析,我们验证了这两种RNA分子在特定肿瘤微环境区域的共定位与协同表达模式,这为揭示其在肿瘤发生发展中的潜在协同调控机制提供了重要线索。

图10. 长链非编码RNA(MALAT1)与信使RNA(PTPN13)的共表达模式,经细胞注释、聚类及HE染色验证
03、宿主与微生物的共表达
时空转录组FFPE产品方案能够同步捕获宿主与微生物的RNA,从而支持宿主-微生物相互作用的联合分析。通过运用Hotspot算法分析,在结核分枝杆菌感染四周后的小鼠肺部模型中,我们识别出97个与病原体基因显著共表达的宿主基因(图11)。KEGG通路分析表明,这些基因在特定组织区域显著富集于免疫相关通路。值得注意的是,巨噬细胞介导的免疫反应与细菌感染高度相关,这一发现与现有免疫学研究结果一致。
 图11. 宿主基因与结核分枝杆菌的共表达模式
图11. 宿主基因与结核分枝杆菌的共表达模式
空间转录组技术(如Stereo-seq)支持的空间基因共表达分析正深刻革新我们对复杂生物系统基因功能的认知。通过整合空间位置信息与转录组数据,该技术使研究者能够系统解析基因表达的空间异质性,尤其在肿瘤亚型分型、lncRNA-mRNA互作网络以及宿主-微生物共表达模式等关键生物学问题中展现出独特优势。我们对三种算法在Stereo-seq数据集中的表现进行了系统评估,揭示了其各自的技术特点:
▪ hdWGCNA:运行速度快的同时展现出更高的基因功能模块一致性;
▪ NeST:运行速度快,且能够精准识别精细解剖结构中的局部共表达模式;
▪ Hotspot:以严格的统计检验(如FDR控制)确保结果可靠性,尤其适合处理数据稀疏的场景。
参数调整建议:
▪ hdWGCNA:降低soft_power参数以增加识别的共表达模块数量。较低的soft_power值可增强网络连通性,从而检测到较弱但具有生物学意义的关联。
▪ NeST:调低hotspot_min_size、hotspot_min_samples或min_cells参数阈值,有助于检测更小或更稀少的共表达模块,避免关键模块因过滤被遗漏。
▪ Hotspot:将fdr_cutoff从0.01调整至0.05以放宽统计严格性,从而增加识别到的模块数量。将模型参数切换至Bernoulli,该模型更适合处理二值或稀疏表达数据。
*本次测评基于研发内部数据,主要覆盖人和小鼠样本,具体效果以实际为准。
参考文献:
1. Walker, B. L., & Nie, Q. (2023). NeST: nested hierarchical structure identification in spatial transcriptomic data. Nature communications, 14(1), 6554.
2. DeTomaso, D., & Yosef, N. (2021). Hotspot identifies informative gene modules across modalities of single-cell genomics. Cell systems, 12(5), 446-456.
3. Morabito, S., Reese, F., Rahimzadeh, N., Miyoshi, E., & Swarup, V. (2023). hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data. Cell reports methods, 3(6).
4. Fan, J., Lu, F., Qin, T., Peng, W., Zhuang, X., Li, Y., ... & Sun, C. (2023). Multiomic analysis of cervical squamous cell carcinoma identifies cellular ecosystems with biological and clinical relevance. Nature genetics, 55(12), 2175-2188.
5. Obayashi et al. (2023) COXPRESdb v8: an animal gene coexpression database navigating from a global view to detailed investigations. Nucleic Acids Research, 51: D80-D87.
内容 | 刘高彤
审核 | 李青娇
