空间转录组学的细胞类型注释在大规模、高分辨率的Stereo-seq数据上面临独特计算挑战。尽管研究者们已提出多种细胞类型注释算法,为特定研究需求选择最优算法仍具挑战性。本文将系统评估不同注释算法的性能和它们在Stereo-seq 数据上的表现。我们的目标是为您提供细胞注释相关的基础性知识,以协助您为自身数据集选择合适的注释算法。
—— 评估结果抢先看 ——
当GPU可用且允许较长运行时间时,鉴于其卓越性能,cell2location通常是首选工具。若需更高计算效率,推荐选择RCTD或Tangram作为替代方案。在分析基因数较少的bin20或cellbin数据时,因Tangram具有更高的注释率,建议优先选用Tangram而非RCTD。若上述工具未能达到预期分析质量,也可以尝试其他算法。
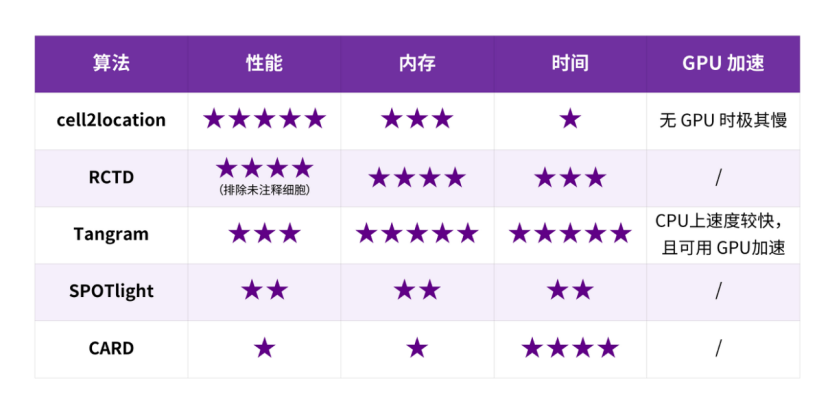
评估算法总结
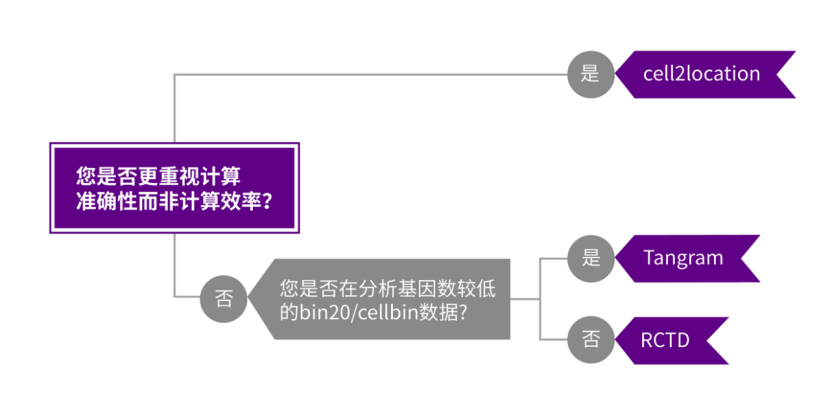
细胞注释算法推荐
细胞类型注释
理解细胞的空间组织及其相互作用,需要确定空间转录组学数据中每个bin或spot的细胞身份。与单细胞转录组学数据不同,基于测序的空间组学数据集通常缺乏单细胞分辨率或表现出有限的基因检出率,这使得注释过程复杂化。
为了应对这一挑战,研究者们已开发出许多算法,将注释过的单细胞参考数据映射到空间位点上,从而将每个位点分解为细胞类型的组合。这个过程称为反卷积(deconvolution),是大多数细胞类型注释算法的核心机制。
注释空间转录组学数据的过程,通常始于筛选参考单细胞数据中各类已注释细胞类型的高表达信息基因,进而生成参考细胞类型的表达矩阵。这个筛选步骤可以由注释算法执行,也可以独立确定。随后,利用数学模型整合参考细胞类型的基因表达矩阵与空间数据,以确定每个空间位置的细胞类型。细胞注释算法的输出通常为每个bin或spot的细胞类型比例,我们可以取一个bin内最丰富的细胞类型来代表它,这称为主要细胞类型鉴定(major cell type identification)(图1)。
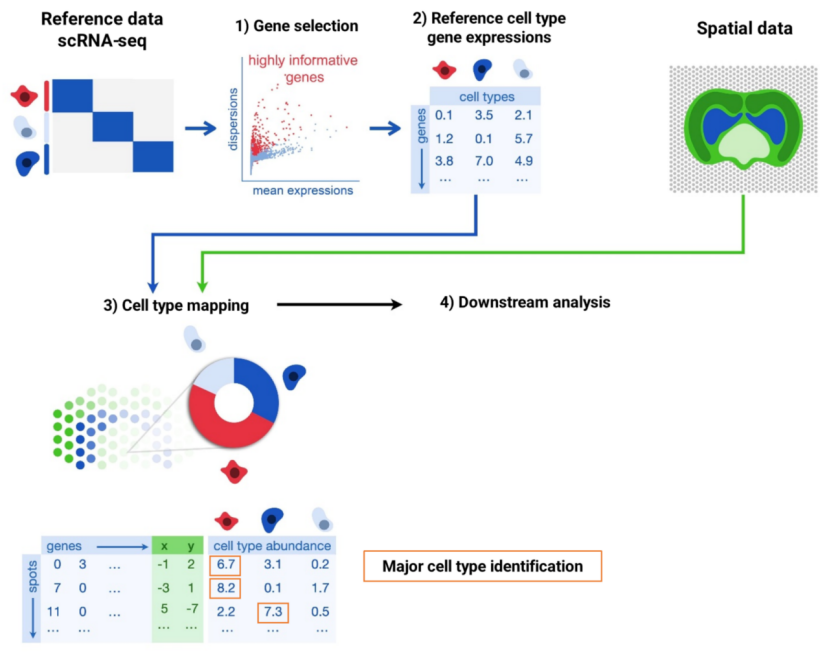
图 1 空间转录组学中的细胞注释
为空间转录组学设计的细胞类型注释算法
利用单细胞参考数据的细胞类型注释算法大致可分为三类:基于概率模型的算法、采用非负矩阵分解(NMF)的算法,以及利用专门设计的损失函数的其他方法。基于现有的基准测试、综述文章1-4以及我们自身的经验,我们选择了5种据报告性能良好的算法进行评估:RCTD5和cell2location6(基于概率模型),SPOTlight7和CARD8(基于NMF),以及Tangram9(基于深度学习)。
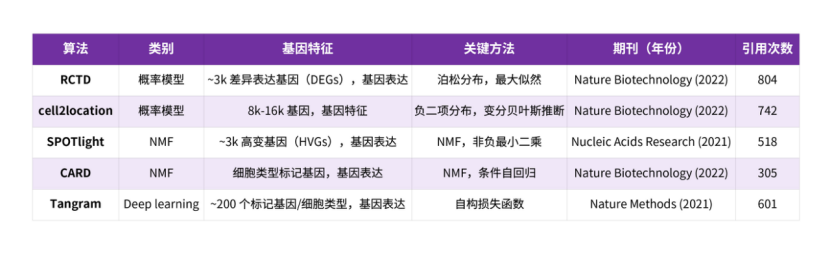
表 1用于评估的选定算法
Stereo-seq 数据上的细胞类型注释算法性能表现
01. 测试的Stereo-seq数据
我们共测试了11个样本,包括小鼠大脑和多种人类肿瘤样本。每个样本测试了cellbin、bin20、bin50和bin100(若可用)。测试数据集覆盖了bin20中基因数从约100到900的不同情况(表2)。

表 2 测试的Stereo-seq数据汇总
02. 评估指标
注释结果的金标准通常通过荧光原位杂交(FISH)技术模拟获得,这在基于测序的空间转录组学数据(如 Stereo-seq)中是不可用的。为了在没有金标准的情况下评估 Stereo-seq 上的算法性能,我们采用了以下 6 个指标:
注释率(Annotation%):表示被成功注释的细胞或bin所占的百分比
PCCm2:预测的细胞类型组成矩阵 (P) 与标记基因表达矩阵 (E) 之间的皮尔逊相关系数 (图2)
标记基因特异性 (Marker Specificity): 标记基因在注释出的细胞类型中的表达 vs. 其他细胞类型中的表达(即量化标记基因热图)
PCCs1: 每种细胞类型中单细胞转录组数据的平均基因表达 (X) 与空间转录组数据的平均基因表达 (Y) 之间的皮尔逊相关系数 (图2)
KLD1: Y 和 X 之间的 KL 散度 (图2)
SSIM1: Y 和 X 之间的结构相似性指数 (图2)
PCCm用于评估bin或spot中细胞类型比例的预测准确性。而标记基因特异性、PCCs、KLD和SSIM则用于衡量每个bin或spot的主要细胞类型的注释准确性和效果。我们认为 PCCm 和标记基因特异性是更可靠的指标,因为它们仅依赖于标记基因。相比之下,PCCs、KLD 和 SSIM 包含了所有检测到的基因,这可能会引入更多噪声,不过我们还是提供这些指标以供参考。
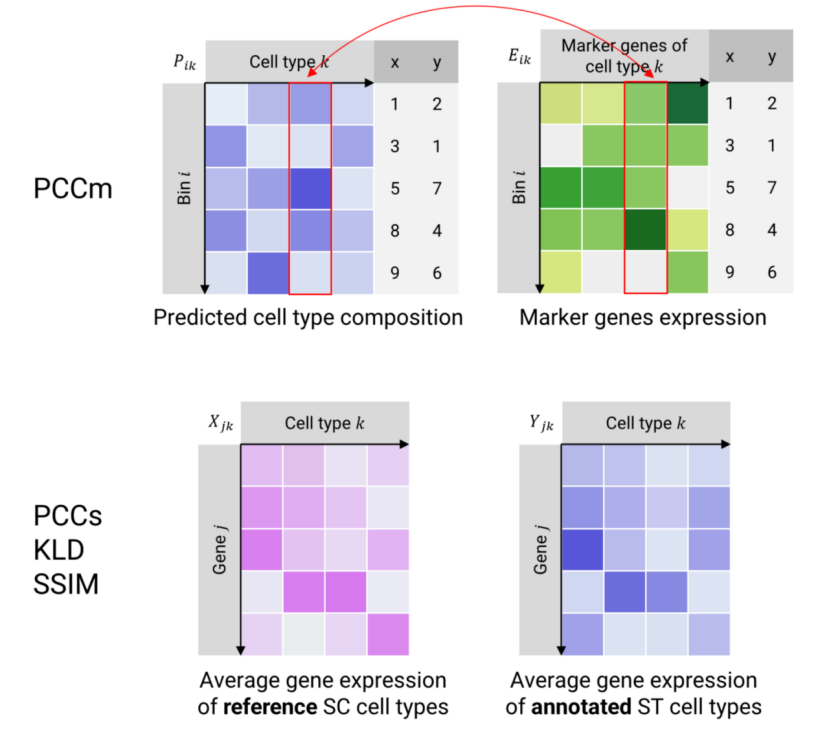
图 2 评估指标示意图
03. Stereo-seq 数据上的性能评估
我们在涵盖小鼠大脑、人结直肠癌、肝癌、乳腺癌和胃癌的 11 个 Stereo-seq 样本上评估了 5 种细胞类型注释算法的性能。
Cell2location 在细胞类型比例预测和主要细胞类型鉴定方面均表现出卓越的性能,获得了总体上最高的 PCCm 和标记基因特异性分数。当排除未注释的bin或细胞时,RCTD 在 bin20 和 cellbin 分析中也得到了接近最优的性能。
Tangram 在 bin20 分析中表现出与 cell2location 和 RCTD 相当的性能。SPOTlight 在 PCCm 和标记基因特异性指标中排名第四。CARD 表现最差,并且由于内存限制无法完成 bin20 或 cellbin 分析(图3)。
此外,RCTD 和 SPOTlight 在 PCCs 和 SSIM 指标上领先,而 Tangram 在评估中得到了最低的 KL 散度分数(图4)。
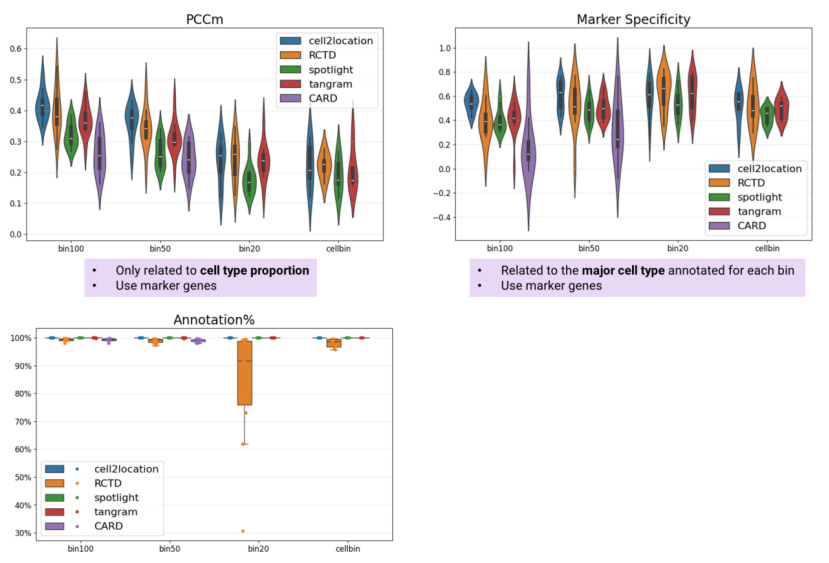
图 3 选定算法的PCCm、Marker Specificity和Annotation%
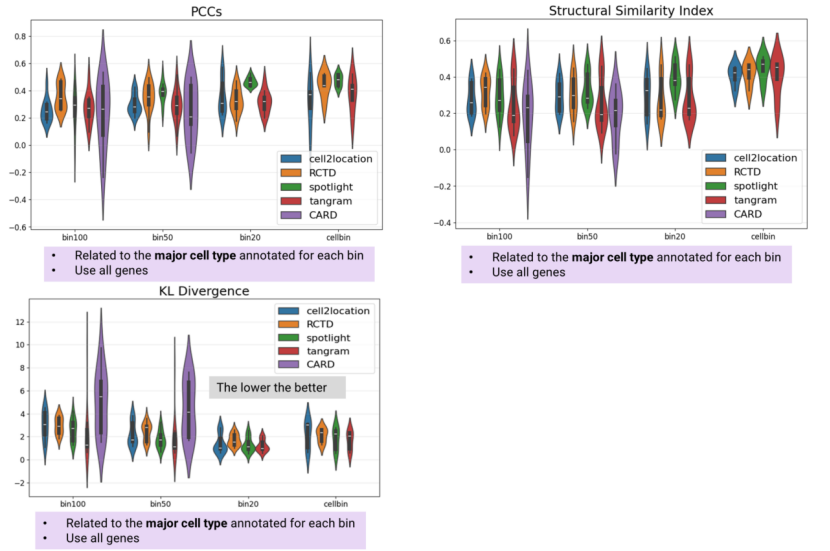
图 4 选定算法的PCCs、SSIM和KLD
04. 时间和内存使用
除了注释准确性,计算效率对于分析大规模 Stereo-seq 数据也至关重要。我们总结了每种算法的时间和内存要求:
Cell2location在需要最长的运行时间,尽管可以使用GPU加速,其内存需求依然较大。相比之下,SPOTlight虽然运行速度比Cell2location快,但其内存消耗更高。RCTD的运行速度显著快于Cell2location,并且在bin100和bin50分辨率下,其速度也明显优于SPOTlight。
此外,RCTD在数据量增长时,内存的消耗并没有显著增加。Tangram在资源需求方面表现最佳,即使没有GPU加速,其运行时间和内存消耗也相对较低。尽管 CARD 也很快,但它消耗了最多的内存,在 500GB 内存限制内无法完成 bin20 和 cellbin 分析(图5)。
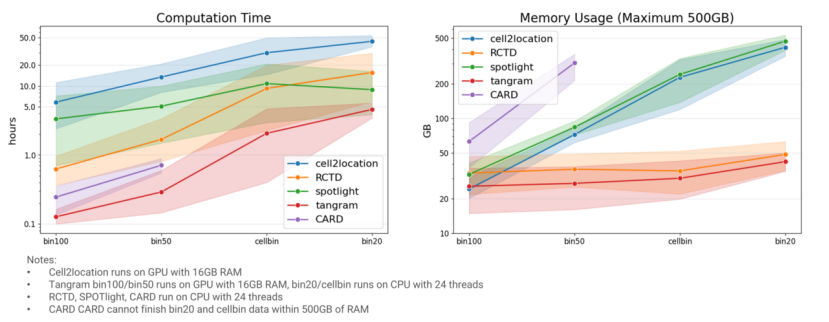
图 5 选定算法的计算资源消耗
05. Bin大小在注释中的影响
我们以这个人结直肠癌样本为例,展示bin大小对细胞类型注释的影响。标记基因热图清晰展示了bin20注释中的对角线模式,相较于bin50和bin100,bin20在主要细胞类型鉴定上更具优势。原因在于,bin50和bin100往往混合了不同细胞类型的信息,而bin20的大小则更接近单个细胞,因此更具区分度。这一观察结果与标记基因特异性从 bin20 到 bin100 的下降趋势一致(图6)。
然而,PCCm 指标与bin大小成反比关系。与标记基因特异性不同,PCCm 评估算法的细胞类型比例预测性能,而不需要识别主要细胞类型。在这种情况下,基因数(随bin大小增加而增加)成为驱动 PCCm 提升的主导因素。这在细胞类型比例图中效果更明显,从 bin20 到 bin100,不同细胞类型的分布变得越来越清晰(图6)。
因此,我们认为 bin20 更适合主要细胞类型鉴定,因为其大小更接近单个细胞。同时,对于低质量样本,我们建议使用 bin100 进行细胞类型比例预测,因为其具有较高的基因数。
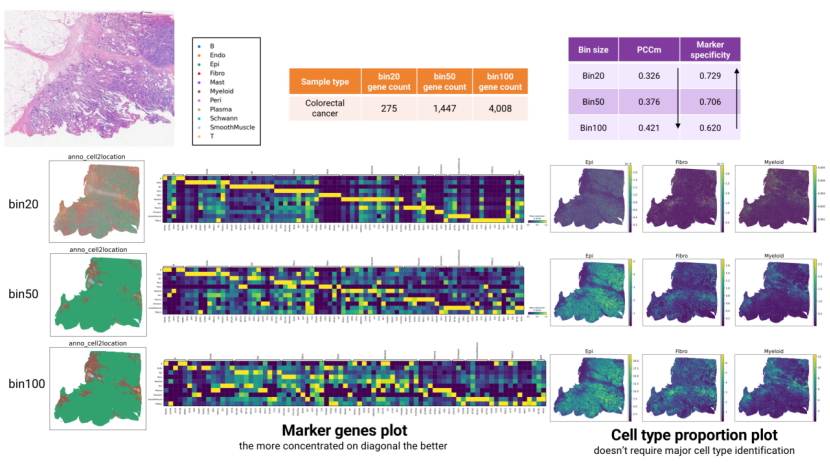
图 6 Bin大小对注释的影响
案例研究
01. 小鼠大脑
对于此类高质量数据集(如这个小鼠大脑样本),所有评估的算法即使在 cellbin 分辨率下也能生成生物学上合理的注释。包括海马体、皮质和丘脑在内的不同结构都能被一致地解析出来。根据 PCCm 和标记基因特异性指标,Cell2location 和 RCTD 取得了最佳性能。关于计算效率,cell2location 的运行时间最长,所需内存超过 150GB。RCTD 和 Tangram 表现出更佳的效率,运行时间少于 3 小时,内存使用量低于 30 GB(图7)。
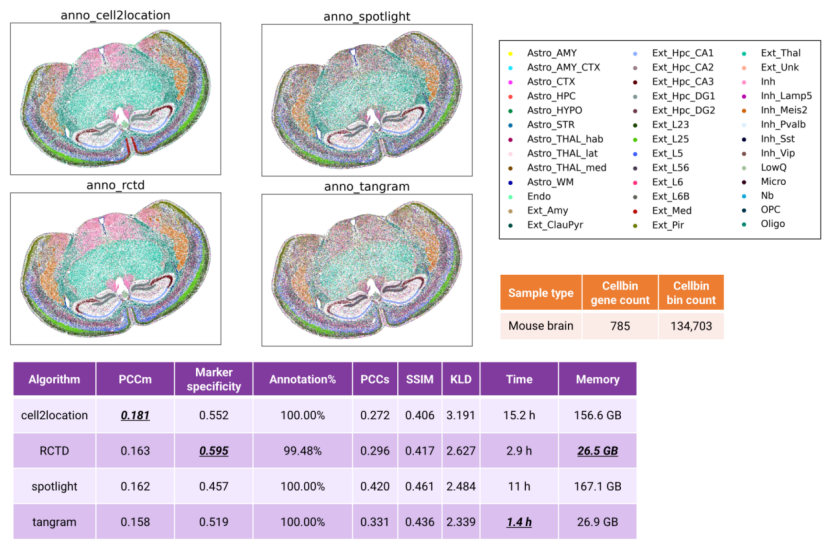
图 7 小鼠脑cellbin数据的细胞注释结果
02. 人结直肠癌中的免疫细胞
此人结直肠癌样本 bin20 数据的中位基因数仅为98,但其中的免疫细胞仍然可以被注释。Cell2location 成功识别了左上角大区域的 B 细胞和 T 细胞,定位了杯状细胞中的 B 细胞聚集区,并检测到平滑肌附近的免疫细胞群。这些结果在主要细胞类型图和细胞类型比例图中均呈现出视觉一致性,标记基因的表达模式也进一步验证了免疫细胞注释的高准确性(图8)。
除 cell2location 外,RCTD、SPOTlight 和 Tangram 也成功注释了这些免疫细胞群。指标评估显示,RCTD 取得了最佳性能,其次是 cell2location 和 Tangram。然而,RCTD 只注释了大约 73% 的bin(图8)。这是因为 RCTD 使用默认参数时会自动过滤低质量数据点。
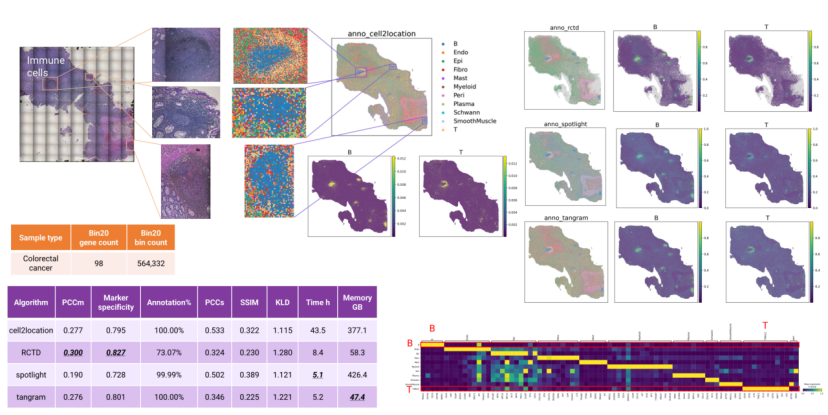
图 8 人结直肠癌bin20数据的细胞注释结果
参考文献
Tao, Q. et al. Benchmarking mapping algorithms for cell-type annotating in mouse brain by integrating single-nucleus RNA-seq and Stereo-seq data. Brief. Bioinform. 25, bbae250 (2024).
Li, H. et al. A comprehensive benchmarking with practical guidelines for cellular deconvolution of spatial transcriptomics. Nat. Commun. 14, 1548 (2023).
Chen, J. et al. A comprehensive comparison on cell-type composition inference for spatial transcriptomics data. Brief. Bioinform. 23, bbac245 (2022).
Li, B. et al. Benchmarking spatial and single-cell transcriptomics integration methods for transcript distribution prediction and cell type deconvolution. Nat. Methods 19, 662–670 (2022).
Cable, D. M. et al. RCTD: Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 40, 517–526 (2022).
Kleshchevnikov, V. et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. 40, 661–671 (2022).
Elosua-Bayes, M., Nieto, P., Mereu, E., Gut, I. & Heyn, H. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 49, e50 (2021).
Ma, Y. & Zhou, X. CARD: Spatially informed cell-type deconvolution for spatial transcriptomics. Nat. Biotechnol. 40, 1349–1359 (2022).
Biancalani, T. et al. Tangram: Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 18, 1352–1362 (2021).
