
学习如何使用核质膜染色图像分割细胞,并得到 Stereo-seq 数据的 CellBin 矩阵,获得更精准的单细胞分辨率的空间转录组解析。
为什么"细胞边界"决定了你的结果可信度
在处理高分辨率的空间组学数据时,我们常常面临一个问题:如何定义一个细胞的边界?
在 Stereo-seq 的标准分析流程中,最常用的方案是“核外扩(Nucleus expansion)”。这种方式就像是用一个饼干模具在复杂的组织面饼上按压——先定位细胞核,再整齐划一地向外扩张若干距离(如 10 个 bin)。
虽然这种几何化的处理方式简单高效,但它往往会让我们与真实的生物学细节失之交臂:
形态失真:真实世界的细胞并非完美的圆,它们可能是长条形的神经元、扁平的内皮细胞或是形态极其不规则的免疫细胞。外扩法不可避免地抹平了这些特征。
信号混杂:在组织密集区,这种基于固定距离的几何外扩难以识别复杂的细胞边界,从而可能将本属于 A 细胞的 mRNA 信号误分配给邻居 B,产生背景噪音。
空间极性丢失: 许多关键的生物过程发生在细胞膜附近。如果边界不准,我们就无法观察到 mRNA 在细胞质内的极性分布。
如果可以引入核质膜多模态图像,利用真实的生物边界来定义细胞范围,我们就可以不再靠猜测推理边界在哪里,而是让图像直接告诉我们边界在哪里。这种“所见即所得”的分割方式,能确保每一个分子都能“各归其位”,从而获得更具生物学意义的 CellBin 矩阵。
学完本教程,你将能够:
用 真实生物边界 替代几何外扩,还原细胞真实形态与极性
跑通 核 + 质 + 膜 三模态深度学习分割,输出可用 Cell Mask
在 StereoMap 中替换 Mask,并回流 SAW
realign流程,重新生成 CellBin 矩阵在 StereoMap 中查看更新后的空间细胞可视化
无论你需要利用 GPU 追求高效率的快速计算,还是需要兼顾通用与轻量化环境的 CPU 逐通道处理,下面都有一条适合你的路径。
准备工作
在开始之前,请确保您已经准备好以下软件工具和文件。
软件工具
示例数据下载
本教程使用一份小鼠肝组织数据作为演示,您可以下载并进行实操。
产品方案:时空转录组FFPE V1.1
染色方案:多模态核质膜染色
芯片号:Y40179K2
核质膜多模态细胞分割,得到 Mask
针对不同的算力条件和数据基础,请选择最适合你的路径:
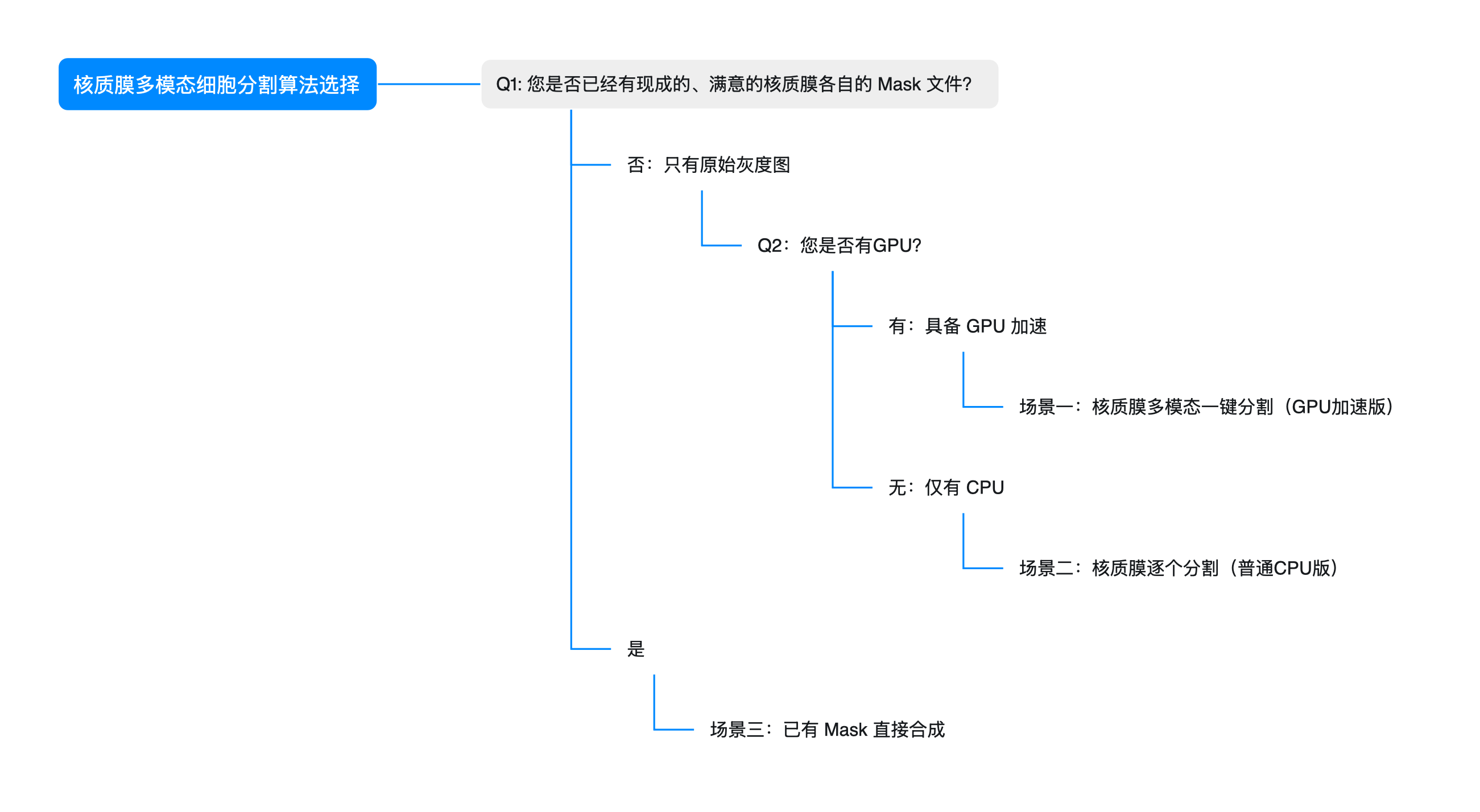
场景一:核质膜多模态一键分割(GPU加速版)
本方案通过整合 核 (Nucleus)、质 (Cytoplasm)、膜 (Membrane) 三路信号,利用深度学习模型精准定义细胞边界。
请确保您的 Conda 环境中已安装 cellbin2,在您的 Terminal 终端输入以下命令,检查您的Cellpose版本是否为4.x.x。
python -c "import cellpose; print(cellpose.version)"
如果不是,先卸载当前的版本。
pip uninstall -y cellpose
然后安装最新的cellpose-4.x.x的版本。
pip install cellpose --upgrade
接下来正式进入分析阶段。复制或直接下载以下脚本为 cellposesam.py。本脚本支持单通道图像自动合成或直接读取多通道彩色图像。
脚本下载:cellposesam.py
由于模型权重较大(约 1.2GB),请提前在服务器终端执行以下命令进行下载:
mkdir -p ./cellseg_weights
wget -P ./cellseg_weights https://github.com/STOmics/cellbin2/releases/download/weights-v1.0.0/cpsam
模式 A:单通道图像接入 (推荐)
适用于 saw count输出的各通道配准图。在核、质、膜三通道齐备的情况下,脚本会自动按 核→蓝、胞质→绿、胞膜→红 的逻辑合成 RGB 图像供模型读取。为兼容不同实验的染色方案,cellposesam.py脚本也支持双通道数据的智能适配。当缺失胞质或胞膜其中任一通道时,脚本将在后台自动执行信号复用,以满足模型的 3 通道输入要求:
核 + 质 场景:仅需输入
--nuc和--cyto。脚本会自动将胞质(Cytoplasm)信号映射至缺失的胞膜(Membrane)通道。核 + 膜 场景:仅需输入
--nuc和--mem。脚本会自动将胞膜(Membrane)信号映射至缺失的胞质(Cytoplasm)通道。
# Estimated time: 1.5 - 2 hours (20k x 20k pixels)
python cellposesam.py \
--nuc ./Y40179K2/outs/image/Y40179K2_DAPI_regist.tif \
--cyto ./Y40179K2/outs/image/Y40179K2_CY5_IF_regist.tif \
--mem ./Y40179K2/outs/image/Y40179K2_TRITC_IF_regist.tif \
-o ./single_channel_cpsam_seg \
-m ./cellseg_weights/cpsam \
-g 1
模式 B:多通道图像接入
适用于已合成好的彩色配准图。
# Estimated time: 1.2 - 1.5 hours (20k x 20k pixels)
python cellposesam.py \
-i ./Y40179K2_merged_RGB.tif \
-o ./multi_channel_cpsam_seg \
-m ./cellseg_weights/cpsam \
-g 1
提示
输入模式强烈建议(针对 16-bit 图像):优先使用【模式 A】。
第三方软件(如 ImageJ)预合并多通道图时,软件为了方便人眼观察,会自动应用直方图拉伸(LUT),导致微弱的细胞边界信号永久丢失。模式 A 直接读取底层物理原图进行无损合并,能确保最高精度的分割结果。(注:若原图设备直出即为 8-bit,则无此差异,可放心使用【模式 B】)。输出路径 (Output Path): 脚本会自动创建输出目录。请确保当前用户具有父目录的写入权限。
分析完成后,您可以在 -o目录下找到生成的 *_cpsam_mask_watershed.tif文件。请将其下载至本地,后续将用于导入 StereoMap 软件以替换原有的 cell masks。
场景二:核质膜逐个分割(普通CPU版)
当您的计算环境没有配置 NVIDIA GPU,或者您需要对 DAPI(核)、CY5(质)、TRITC(膜)通道分别应用不同的分割策略时,请参考本场景。本方案分为分割推理和融合两个阶段。
第一阶段:多通道独立分割
在该阶段,我们将针对不同染色通道,利用最适合的算法引擎生成原始掩码(Mask)图像。
胞核分割:
# Estimated time: 1 hour (20k x 20k pixels)
python cell_segmentor.py \
-i ./Y40179K2/outs/image/Y40179K2_DAPI_regist.tif \
-o ./cell_seg_masks \
-p ./cellseg_weights \
-s dapi \
-g -1脚本下载:cell_segmentor.py
使用 CellBin2 内置模型处理 DAPI 通道,定位细胞核心。
算法引擎: BCDU / UNet (CellBin2 Built-in)
胞质与胞膜分割:
使用 Cellpose 引擎分别提取质膜轮廓。
算法引擎: Cellpose (Model: cyto2)
# Estimated time: 1 hour for each image (20k x 20k pixels)
python cellpose_segmentor.py \
-i ./Y40179K2/outs/image/Y40179K2_CY5_IF_regist.tif \
-o ./cell_seg_masks \
-m ./cellseg_weights \
-n cyto2 \
-g -1
python cellpose_segmentor.py \
-i ./Y40179K2/outs/image/Y40179K2_TRITC_IF_regist.tif \
-o ./cell_seg_masks \
-m ./cellseg_weights \
-n cyto2 \
-g -1
第二阶段:多源轮廓集成与优选
在该阶段,脚本会对比胞核、胞质、胞膜各模态生成的初步轮廓。针对每一颗细胞,算法将动态评估并筛选出最能反映其实际形态的边界来源,最终集成导出为完整的 Cell Mask。
为兼容不同实验的染色方案,脚本支持双通道数据的适配:胞核 Mask 为严格必选项,当缺失胞质或胞膜其中任一通道时,只需输入已有的参数,脚本即可自动切换对应的融合策略。
请复制或下载以下脚本为 multimodal_cell_merge.py。
接下来运行融合命令:
# Estimated time: 0.5 hours
python multimodal_cell_merge.py \
--nuc ./cell_seg_masks/Y40179K2_DAPI_regist_v3_mask.tif \
--cyto ./cell_seg_masks/Y40179K2_CY5_IF_regist_cellpose_mask.tif \
--mem ./cell_seg_masks/Y40179K2_TRITC_IF_regist_cellpose_mask.tif \
-o ./cell_seg_masks \
-d 10
运行成功后,您可以在 ./cell_seg_masks目录下找到生成的 final_cell_mask.tif文件。请将其下载至本地,后续将用于导入 StereoMap 软件以替换原有的分割掩码。
场景三:已有 Mask 直接合成
该场景适用于您已经拥有了独立的核、质、膜 Mask 文件(例如从第三方软件导出或自训练模型生成等),仅需要将其整合为一张单细胞掩码。
请确保您的工作目录下已保存场景二中提供的 multimodal_cell_merge.py 脚本。直接调用脚本对您的既有 Mask 进行逻辑合并:
# Estimated time: 0.5 hours
python multimodal_cell_merge.py \
--nuc ./your_masks/existing_nuclei_mask.tif \
--cyto ./your_masks/existing_cytoplasm_mask.tif \
--mem ./your_masks/existing_membrane_mask.tif \
-o ./cell_seg_masks \
-d 10
重要提醒:坐标对齐
融合脚本仅负责 Mask 融合,不负责图像配准。请确保输入的三个 Mask 已经基于同一坐标系完成了对齐,以便后续使用。
导入多模态细胞分割 Mask
完成多模态 Mask 的生成后,即可将其导入 StereoMap 软件进行后续的空间可视化与矩阵计算。
启动 StereoMap,进入 Image Processing 模块。在图像类型(Image Type)下拉菜单中,选择 DAPI+mIF (Nuclei-staining image + immunofluorescence image) 染色模式。
根据您的具体情况,选择对应的文件格式进行图像上传:
- 推荐上传
.stereo文件:如果您在 SAW 流程中已启用自动图像分析并成功生成结果,请直接上传该文件(其内部已包含完整的空间配准信息)。 - 备选上传
.tar.gz图像包:如果您的图像在前期 QC 阶段未通过,或者在运行 SAW 分析时完全没有使用图像参数,请上传原始的图像压缩包。

- 推荐上传
当上传的是
.stereo文件且配准无需调整时,可在 Step 2:Image Registration 中直接点击 Next,进入后续步骤。
空间坐标的一致性至关重要!
如果您在此步骤中手动调整了配准,这意味着图像坐标已发生改变。您必须使用新导出的
*_regist.tif文件,重新运行第一步的“多模态细胞分割”脚本以生成新的 Mask。在 Step 2 手动导入矩阵并进行配准(Morphology 或 Feature Point);
完成后,连续点击 Step 3 和 Step 4 的 Next,进入 Step 5:Export;
导出后将在目标目录生成
*_regist.tif文件。进入 Step 4:Cell Segmentation 模块,点击 Segmentation mask 下拉框
点击 Custom 下 Add mask 选项右侧的“+”添加 mask。、
在弹出的文件选择窗口中,选择第一步生成的多模态细胞分割 TIFF mask。

成功导入后,Mask 将以“细胞轮廓”的形式叠加显示在画布上,底层同步映射配准后的显微镜图像。在此界面您可以自由放大缩小画布,仔细检查分割效果。也可利用手动编辑工具修饰局部分割边界。任何新增或修改的操作,软件都会自动重新计算当前的细胞总数。若对当前Mask的整体效果不满意,直接再次选择并上传新的 TIFF mask,即可一键覆盖并替换。

Mask 替换完成后,进入Step 5,选择目标路径导出更新结果。更新后的结果文件为替换了 mask 的图像
.tar.gz包。

重新计算 CellBin 矩阵
这是最后一步,也是见证结果的一步:把更新 Mask 后的图像回流 SAW,重新生成基于真实边界的 CellBin 矩阵。
将 StereoMap 在本地导出的图像
.tar.gz包上传至部署了 SAW 的服务器,然后将其输入至 SAW 的realign流程。该流程将基于多模态细胞分割结果重新提取 CellBin 矩阵。# 输入更新后的图像 .tar.gz 运行 saw realign流程,重新提取 CellBin 矩阵
saw realign \
--id=Y40179K2_realign \
--sn=Y40179K2 \
--count-data=Y40179K2_Mouse_Kidney \
--realigned-image-tar=Y40179K2_SC_20260415_153736_4.3.0.tar.gz \
--threads-num=24需要注意的是,
--count-data参数应传入第 1 步中上传.stereo文件时所使用的 SAWcount分析结果目录。运行完成后,
outs目录中的.cellbin*.h5ad和.cellbin.gef文件即为基于多模态分割结果重新生成的细胞矩阵。同时,.html网页报告中的细胞统计与聚类结果,也根据这些更新后的矩阵重新计算。下载并解压
visualization.tar.gz文件,随后将其导入 StereoMap 的 Visual Explore 模块,即可直观查看基于全新 CellBin 矩阵的空间可视化效果。

看见真实边界,看见真实生物学
回到我们开头的问题:怎么定义一个细胞的边界?
核外扩给出的是一个方便的近似;多模态分割给出的是图像里真实存在的轮廓。从形态保真、信号归属到空间极性,每一次边界精度提升,都会沿着下游聚类、细胞类型注释、空间通讯分析一路传递下去。
现在你可以:
🔬 下载示例数据,用小鼠肝组织跑通完整流程:获取示例数据
🛠️ 拉取 cellbin2 多模态分支,开始你的第一次三模态分割:cellbin2 (feature/multimodal分支)
📚 关注本系列,后续我们将持续推出更多实操指南。
附录:脚本与权重速查
版本说明:本教程基于 SAW ≥ 8.3.0、StereoMap ≥ 4.3.0、cellbin2 feature/multimodal 分支、Cellpose 4.x.x、Python 3.8 验证。